Abstract
Cyclopropenes as substrates entered the field of gold catalysis in 2008 and have proven to be valuable partners in a variety of gold-catalyzed reactions. The different contributions in this growing research area are summarized in this review.

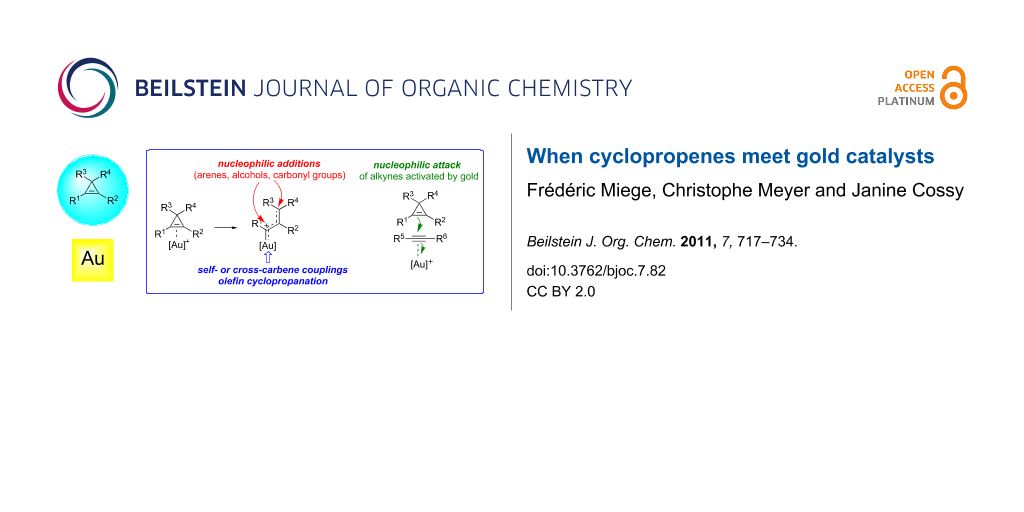
Graphical Abstract
Review
Introduction
Homogeneous gold catalysis has become a particularly active research area over the last decade. The ability of gold catalysts to act as potent carbophilic Lewis acids and hence to chemoselectively activate π bonds towards nucleophilic attack is now well-established and has found many impressive applications for the formation of C–C or C–heteroatom bonds [1-14]. Whereas alkynes, alkenes and allenes have been widely used as substrates or partners in gold-catalyzed reactions, it was only rather recently, in 2008, that cyclopropenes entered the field of gold catalysis despite their well-known high and versatile reactivity in transition metal-catalyzed reactions [15].
As has been observed with other transition metals, the reactivity of cyclopropenes A in gold-catalyzed reactions is essentially (but not exclusively) related to their ability to act as ligands for π-acidic gold complexes, and hence, to undergo subsequent ring-opening to produce an organogold species that can be viewed as a hybrid between a gold-stabilized allylic carbocation B and a gold carbene C. The organogold carbenoid species generated by the ring-opening of cyclopropenes can participate in a variety of reaction types such as nucleophilic addition with, e.g., alcohols, arenes or carbonyl groups, undergo self- or cross-carbene couplings and bring about the cyclopropanation of olefins. The first of these reaction types is often considered to be representative of cationic intermediates whereas the other two are best ascribed to carbene-like reactivity, although this distinction is artificial. Alternatively, cyclopropenes can also behave as nucleophiles and attack other functional groups that are more readily activated by gold complexes, such as alkynes (Scheme 1) [16-26].
![[1860-5397-7-82-i1]](/bjoc/content/inline/1860-5397-7-82-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General reactivity of cyclopropenes in the presence of gold catalysts.
Scheme 1: General reactivity of cyclopropenes in the presence of gold catalysts.
This review illustrates the different aspects of the reactivity of cyclopropenes in the presence of gold catalysts and covers the contributions in this field up to February 2011.
Besides their implication in several gold-catalyzed reactions, cyclopropenes have also served as substrates in order to gain insight into the gold-carbon order in the so-called organogold carbenoids. In the broad repertoire of gold-catalyzed organic transformations, gold-stabilized carbocations or, more often gold carbenes, can be found as intermediates in proposed mechanistic pathways, but the true nature of the organogold species had been a matter of debate [27].
Structural considerations: Gold-stabilized carbocations or gold carbenes?
In 2008, Fürstner et al. took advantage of the ring-opening of 3,3-disubstituted cyclopropenes to generate organogold species and characterize them by NMR spectroscopy [16]. Whereas, 3,3-diphenylcyclopropene (1) or 3,3-dimethylcyclopropene (2) did not generate a defined organogold species upon treatment with Gagosz’s complex [(Ph3P)AuNTf2] [28] (CD2Cl2, −78 °C) due to rapid oligomerization, the cyclopropenone acetal 3 gave an organogold species whose NMR spectroscopic data corresponds to the carbocationic structure (Z)-4a. Upon raising the temperature, organogold (Z)-4a was found to isomerize into its geometric isomer (E)-4a. Switching to more electron-donating phosphine ligands such as PMe3 or PCy3, also led to the organogold species 4b and 4c, respectively, possessing a dioxacarbenium structure, with the predominance of the E geometric isomers already at −80 °C. The observed data point towards a high degree of double bond character for the C1–C2 bond, and not the C2–C3 bond, in the organogold species generated by ring-opening of cyclopropenone acetal 3, with a marginal contribution of the carbene form 5. The magnitude of the rotational barrier around the C2–C3 bond for 4a (<30 kJ·mol−1) was in agreement with this result. In the case of the less stable organogold species (Z)-7, generated from 3,3-dimethoxycyclopropene (6) using [(Me3P)AuNTf2], the broadening of the NMR signals indicated a more restricted rotation around the C2–C3 bond at −80 °C, but the rotation barrier estimated to be 46 ± 1 kJ·mol–1 was still comparable in magnitude to rotation around a sterically hindered σ bond (such as in hexachloroethane) (Scheme 2) [16].
![[1860-5397-7-82-i2]](/bjoc/content/inline/1860-5397-7-82-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cationic organogold species generated from cyclopropenone acetals.
Scheme 2: Cationic organogold species generated from cyclopropenone acetals.
These experiments appeared to be useful for the determination of the cationic or carbenic nature of organogold intermediates, but the presence of the two oxygen atoms in cyclopropenone acetals unavoidably led to more favorable cationic forms and hence cannot provide a general answer.
Using the M06 functional of DFT, Toste et al. calculated rotational barriers for (Z)-4a and (Z)-7 and the results were found to be in agreement with those previously obtained experimentally by Fürstner et al. Thus, with this validated computational method, the barriers to bond rotation in (metal free) 3,3-disubstituted allyl cations, and in the corresponding (Me3P)Au-substituted organogold species, were calculated. Unlike in the case of the allylic cation bearing an acetal moiety at C3, incorporation of the gold center at C1 in the 3,3-dimethyl substituted allylic cation raised the rotation barrier considerably to 94 kJ·mol−1, and hence the latter species should be regarded more as a gold carbene (Scheme 3) [17].
![[1860-5397-7-82-i3]](/bjoc/content/inline/1860-5397-7-82-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Rotation barriers around the C2–C3 bond (M06 DFT calculations).
Scheme 3: Rotation barriers around the C2–C3 bond (M06 DFT calculations).
The bond distances and natural atomic charges were calculated for a series of 3,3-disubstituted allylic cations, bearing an acetal, two methyl or two carbomethoxy groups, as well as for their (trimethylphosphine)gold-substituted counterparts. The results indicate that a secondary gold-substituted carbocation (at C1) is as stable as a tertiary dimethyl-substituted carbocation (at C3) and that the magnitude of stabilization from the gold moiety increases with increasing electrophilicity of the allylic cation. Toste et al. investigated the effect of the ligand on the structure of gold-substituted 3,3-dimethyl allyl cations of type D. Increasing trans σ-donation from the ligand and strongly π-acidic ligands such as phosphites (decreasing back π-donation from gold to C1) led to a longer C1–Au bond and hence a more carbocation-like character for the organogold species. By contrast, those ligands that increase gold-to-C1 back π-donation or decrease C1-to-gold σ-donation will induce a shorter C1–Au bond and a carbene-like reactivity (Scheme 4) [17].
![[1860-5397-7-82-i4]](/bjoc/content/inline/1860-5397-7-82-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Au–C1 bond length in organogold species of type D.
Scheme 4: Au–C1 bond length in organogold species of type D.
These studies highlighted the tremendous influence of the substitution pattern and the ancillary ligand on the nature of bonding in cationic gold-stabilized intermediates. Interestingly, the organogold species investigated in these computational studies are precisely those that can be generated by the ring-opening of cyclopropenes in the presence of gold complexes. Indeed, as it will be illustrated later in this review, these structural effects were found to have important consequences in terms of reactivity in the case of intermolecular olefin cyclopropanation promoted by gold carbenes generated from cyclopropenes.
In fact, the first reports on gold-catalyzed reactions involving cyclopropenes appeared in the literature before these structural investigations were carried out. In the following presentation of the different chemical transformations involving cyclopropenes, either one of the two forms (i.e., an allylic gold cation or carbene) will be drawn in the mechanistic pathway. In general, little information is available on the modulation and tuning of the reactivity by the choice of the gold ligand.
Nucleophilic addition to gold-stabilized allylic cations generated from cyclopropenes
Intermolecular addition of oxygen nucleophiles
In 2008, Lee et al. reported several gold-catalyzed reactions involving cyclopropenes among which the addition of alcohols to 3-methyl-3-nonylcyclopropene (8) was investigated in detail [18]. A variety of primary alcohols reacted with cyclopropene 8 in the presence of either in situ generated [(Ph3P)AuOTf] or [(Ph3P)AuNTf2] (5 mol %) to afford the corresponding tert-allylic ethers 9a–9f with very high regioselectivity (>99%). Other catalysts such as AuCl3 or Rh2(OAc)4 provided mixtures of compounds containing traces of allylic ethers 9 and 9’ and mostly oxidation products (vide infra, enals 16 and 17). AgOTf was less efficient and led to an incomplete conversion whereas no reaction took place with TfOH. Gagosz’s catalyst was in general more efficient than [(Ph3P)AuOTf] and also allowed iPrOH to be used as a nucleophile to provide ether 9g (70%), however, tertiary alcohols did not react under these conditions. Water in the presence of t-BuOH as co-solvent acted as a nucleophile, but the corresponding tertiary alcohol 9i was isolated in only modest yield (34%) (Scheme 5) [18]. Additional results were subsequently reported by Lee et al. in a full article in 2010 [19]. Due to their lower nucleophilic character compared to alcohols, phenols could not be used. With the optically pure chiral alcohol (R)-PhMeCHCH2OH as a nucleophile, the reaction was not diastereoselective and led to the tertiary allylic ether 9k (65%) as a 1:1 mixture of diastereomers. An unprotected primary and tertiary 1,3-diol reacted chemoselectively with the primary alcohol to furnish monoether 9l (58%). Addition of neopentyl glycol led to a 1:1 mixture of regioisomeric monoethers 9m and 9’m in modest yield (32%) due to the competitive formation of oligomeric by-products. The regioselectivity was found to be highly sensitive to temperature since the tertiary monoether 9m was selectively obtained (9m/9’m > 99:1) (33%) when the reaction was carried out at 10 °C (Scheme 5) [18,19].
![[1860-5397-7-82-i5]](/bjoc/content/inline/1860-5397-7-82-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Gold-catalyzed addition of alcohols or water to cyclopropene 8.
Scheme 5: Gold-catalyzed addition of alcohols or water to cyclopropene 8.
The reaction was successfully extended to a variety of 3,3-disubstituted cyclopropenes (3-methyl-3-benzylcyclopropene, spiro[2.5]oct-1-ene, 3-benzyl-3-isopropylcyclopropene, 3-tert-butyl-3-methylcyclopropene) and the corresponding tertiary allylic ethers were always obtained with high regioselectivities (92:8 to >99:1). However, when 3-methyl-3-phenylcyclopropene (10) was used as the substrate the regioselectivity was altered in some cases. With n-BuOH as a nucleophile, a 1:1 regioisomeric mixture of allylic ethers 11a and 11’a was obtained under the previously used reaction conditions. By lowering the temperature to 10 °C and increasing the quantity of n-BuOH (15 equiv), the tertiary allylic ether 11a (65%) was obtained regioselectively (11a/11’a > 99:1). Curiously, a complete switch of the regioselectivity took place when phenethyl alcohol was employed as a nucleophile, since in this case the primary allylic ether 11’b (65%) was obtained (11b/11’b > 1:99) (Scheme 6) [19].
![[1860-5397-7-82-i6]](/bjoc/content/inline/1860-5397-7-82-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Gold-catalyzed addition of alcohols to cyclopropene 10.
Scheme 6: Gold-catalyzed addition of alcohols to cyclopropene 10.
The formation of the tert-allylic ethers 9 can be explained by the regioselective attack of the alcohol at C3 on the organogold species 12, generated by electrophilic ring-opening of cyclopropene 8, followed by protodeauration of the resulting vinyl gold species 13. Using CD3OD as a nucleophile effectively led to 90% deuterium incorporation at C1 and formation of a mixture of geometric isomers (Scheme 7) [18,19].
![[1860-5397-7-82-i7]](/bjoc/content/inline/1860-5397-7-82-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Mechanism of the gold-catalyzed addition of alcohols to cyclopropenes.
Scheme 7: Mechanism of the gold-catalyzed addition of alcohols to cyclopropenes.
Interestingly, an excess of the alcohol (6 equiv) was crucial to achieve high regioselectivities. If the quantity of EtOH was reduced (1 equiv) a 2:1 mixture of the corresponding regioisomeric allylic ethers 9a and 9’a was obtained, however, the addition of a protic additive [t-BuOH (5 equiv)] restored the high regioselectivity (>99:1) [18,19]. Lee and Hadfield demonstrated that the use of an excess of methanol retarded the isomerization of the tertiary allylic ether 9b into the primary allylic isomer 9’b, which is also catalyzed by the gold complex [29] (Scheme 8). The isomerization was also found to be catalyst dependent and did not operate in the presence of the NHC–gold complex [(IPr)AuOTf]. Thus, when cyclopropene 8 was treated with a stoichiometric quantity of EtOH in the presence of the latter catalyst (5 mol %), the tertiary allylic ether 9a was obtained with high regioselectivity (>99:1), but the yield (51%) was not as high as with Gagosz’s catalyst (83%). Lee and Hadfield took advantage of these findings to develop the regioselective addition of alcohols (used in excess) to allenes such as 14 catalyzed by [(IPr)AuOTf] (10 mol %) to produce the tert-allylic ethers 15 as the kinetic products (Scheme 8) [29].
![[1860-5397-7-82-i8]](/bjoc/content/inline/1860-5397-7-82-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of tert-allylic ethers from cyclopropenes and allenes.
Scheme 8: Synthesis of tert-allylic ethers from cyclopropenes and allenes.
During their studies on the addition of alcohols to cyclopropenes, Lee et al. also reported one example of oxidation of the gold carbene intermediate 12, resulting from the electrophilic ring-opening of 3-methyl-3-nonylcyclopropene (8), with diphenylsulfoxide [30]. The reaction proceeds by nucleophilic attack of diphenylsulfoxide at C1 followed by elimination of diphenylsulfide to afford a 55:45 mixture of the E and Z enals 16 and 17, respectively (66%) (Scheme 9) [18,19].
![[1860-5397-7-82-i9]](/bjoc/content/inline/1860-5397-7-82-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Oxidation of the intermediate gold–carbene with diphenylsulfoxide.
Scheme 9: Oxidation of the intermediate gold–carbene with diphenylsulfoxide.
Other examples of nucleophilic attack on organogold species resulting from the ring-opening of cyclopropenes in the presence of gold complexes involve intramolecular Friedel–Crafts reactions and the addition of carbonyl groups.
Intramolecular Friedel–Crafts reactions
In the context of their studies on the Lewis acid-catalyzed rearrangement of strained three-membered ring hydrocarbons, such as methylenecyclopropanes and vinylidenecyclopropanes, Shi et al. investigated the behaviour of 1-(2,2-diarylvinyl)-2-phenylcyclopropenes in the presence of gold catalysts [20]. Upon treatment with [(Ph3P)AuOTf], vinylcyclopropene 18 was found to produce a mixture of regioisomeric indenes 19 and 20 in a 75:25 ratio (99%). The use of AgOTf alone led to the isomeric substituted naphthalene 21 as the sole product. Shi et al. had previously demonstrated that indene 20 and naphthalene 21 could be selectively formed using Cu(OTf)2 and BF3·OEt2 as catalysts, respectively, thereby highlighting the complementarities of the different electrophilic activators [31]. Since AgOTf and BF3·OEt2 led to the same naphthalene product 21, the authors suspected that traces of the Brønsted acid (HOTf) present in the silver salt may be the actual catalyst and may also modify the regioselectivity observed in the gold-catalyzed reaction. Thus, several basic additives were screened and it was found that DBU not only inhibited the isomerization of vinylcyclopropene 18 in the presence of AgOTf, but also led to a completely regioselective gold-catalyzed process to afford indene 19 as the sole reaction product (97%) (Scheme 10) [20].
![[1860-5397-7-82-i10]](/bjoc/content/inline/1860-5397-7-82-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Gold, copper and Lewis acid-catalyzed reactions of cyclopropene 18.
Scheme 10: Gold, copper and Lewis acid-catalyzed reactions of cyclopropene 18.
Upon electrophilic activation, vinylcyclopropene 18 can give rise to two regioisomeric cyclopropyl cations 22 and 23. It is worth noting that consideration of these two cation species is only helpful to understand the observed regioselectivities, though they may not be actually involved as intermediates during the ring-opening of cyclopropenes in the presence of electrophilic transition metal complexes. Shi et al. initially suggested that the formation of 22 preferentially occurs with a rather bulky electrophile such as Cu(OTf)2 to avoid repulsion with the aryl group at C2. Conversely, a Brønsted acid (generated by reaction of BF3·OEt2 with traces of water) should favour the formation of the more stable cyclopropyl cation 23. Afterwards, ring-opening and intramolecular Friedel–Crafts reactions should enable the formation of indene 20 or naphthalene 21. With the gold catalyst, the formation of indene 19 indicated that electrophilic activation of the cyclopropene 18 also occurred at C2 to afford, after ring-opening, the gold-stabilized allylic cation 24. However, in contrast to the acid-catalyzed reaction, subsequent intramolecular Friedel–Crafts cyclization occurred by nucleophilic attack by the phenyl group (at C2) on the organogold species at C3, followed by protodeauration (Scheme 11) [20,31].
![[1860-5397-7-82-i11]](/bjoc/content/inline/1860-5397-7-82-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Mechanism of the Lewis acid-catalyzed reactions of cyclopropene 18.
Scheme 11: Mechanism of the Lewis acid-catalyzed reactions of cyclopropene 18.
The reaction was generalized with a series of 3,3-disubstituted-1-(2,2-diarylvinyl)-2-arylcyclopropenes of general formula 25. The catalyst [(Ph3P)AuSbF6] was found to provide better results than [(Ph3P)AuOTf] for substrates having electron-withdrawing substituents on the benzene rings. The corresponding indenes 26 were obtained in good to excellent yields (85–99%) under the previously optimized conditions (Scheme 12) [20].
![[1860-5397-7-82-i12]](/bjoc/content/inline/1860-5397-7-82-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Gold-catalyzed rearrangement of vinylcyclopropenes 25.
Scheme 12: Gold-catalyzed rearrangement of vinylcyclopropenes 25.
In the absence of substituents at C3 (R3 = R4 = H), or when a single substituent was attached to this carbon (R3 = Me, R4 = H), the reaction led to a complex mixture of products. The authors attributed these results to the formation of less stable carbocations at C3 (primary or secondary, respectively).
Other examples of gold-catalyzed isomerization of cyclopropenes that involve a Friedel–Crafts cyclization have been reported. In 2009, Wang et al. demonstrated that [(Ph3P)AuOTf] could smoothly catalyze the isomerization of a variety of 3-substituted 1,2,3-triphenylcyclopropenes 27 into 3-substituted 1,2-diphenyl-1H-indenes 28 [21]. The rearrangement occurred rapidly (20–40 min) for substrates 27a–27e and indenes 28a–28e were obtained in excellent yields (97–99%). A phenylethynyl group could be present at C3, but the rearrangement of substrate 27f proceeded slowly (rt, 6 h) and gave indene 28f in only a moderate yield (54%) together with an unknown by-product, presumably because the alkyne competes with the cyclopropene for coordination to the gold catalyst (Scheme 13) [21,26].
![[1860-5397-7-82-i13]](/bjoc/content/inline/1860-5397-7-82-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Gold-catalyzed rearrangement of cyclopropenes 27 to indenes 28.
Scheme 13: Gold-catalyzed rearrangement of cyclopropenes 27 to indenes 28.
The rearrangement of 27a to 28a (95%) had been previously reported by Müller et al. using rhodium(II) perfluorobutyrate as a catalyst (1 mol %, C6H6, reflux, 48 h) [32], whereas Padwa et al. showed that the isomerization of 27b to 28b was quantitatively catalyzed by AgClO4 (2 mol %, C6H6, rt) [33].
Wang et al. also examined the behaviour of 3-arylcyclopropenes bearing a protected hydroxymethyl group at C3: only acetates 29 underwent clean conversion to 1-methylene-2-substituted-1H-indenes 30 [21]. The yields were improved by the addition of DBU once the rearrangement was complete. For substrates 29 possessing an unsymmetrically substituted endocyclic olefin, it is worth noting that electrophilic activation of the cyclopropene occurred regioselectively to produce the organogold species 31 (formally resulting from the ring-opening of a secondary benzylic cyclopropyl cation). The gold carbene 31 was captured by the aromatic group at C3 via an intramolecular Friedel–Crafts reaction. Subsequent elimination of AcOH from compound 33 then delivered methylene indene 30 (Scheme 14) [21].
![[1860-5397-7-82-i14]](/bjoc/content/inline/1860-5397-7-82-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Gold-catalyzed rearrangement of cyclopropenes 29 to indenes 30.
Scheme 14: Gold-catalyzed rearrangement of cyclopropenes 29 to indenes 30.
Other gold-catalyzed rearrangements of cyclopropenes that proceed through ring-opening and intramolecular Friedel–Crafts cyclization have been studied using 3-aryl-cyclopropene-3-carboxylates. However, for these latter substrates, the carbonyl group can also play the role of a nucleophile and compete with the aryl group.
Nucleophilic addition of carbonyl groups in competition with Friedel–Crafts reactions
Besides the gold-catalyzed intermolecular addition of alcohols to cyclopropenes, Lee et al. investigated the behaviour of methyl 3-arylcyclopropen-2-yl carboxylates to ascertain whether the organogold species resulting from the ring-opening in the presence of [(Ph3P)AuOTf] (10 mol %) would be trapped in an intramolecular fashion, either by the oxygen atom of the carbonyl group, or by the phenyl group [18]. For cyclopropene 34a possessing an unsubstituted endocyclic alkene, heating in toluene (80 °C, 18 h) was required and the reaction afforded two products: Furanone 35a (52%) and indene 36a (20%). The former compound arose from intramolecular trapping of the intermediate organogold species 37a by the carbonyl group of the ester at C3, followed by hydrolysis of the resulting α-methoxyfuran 38a. A similar result was reported by Wang et al. [21]. Indene 36a is, as previously mentioned, the product resulting from an intramolecular Friedel–Crafts reaction (Scheme 15).
![[1860-5397-7-82-i15]](/bjoc/content/inline/1860-5397-7-82-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Gold-catalyzed rearrangement of cyclopropenyl ester 34a.
Scheme 15: Gold-catalyzed rearrangement of cyclopropenyl ester 34a.
For unsymmetrical cyclopropenes 34b–34d possessing a trisubstituted endocyclic double bond, the rearrangement took place at rt and invariably led to mixtures of furanones 35b–35d, and mixtures of the inseparable regioisomeric indenes 36b–36d and 36’b–36’d. Electrophilic activation and ring-opening of cyclopropenes 34 favored the formation of the organogold species 37b–37d. Furanones 35b–35d and indenes 36b–36d result from nucleophilic attack on these latter intermediates at C1 by the carbonyl or the phenyl group, respectively. By contrast, the regioisomeric indenes 36’b–36’d would arise from the initial formation of organogold species 37’b–37’d and subsequent Friedel–Crafts cyclization (Scheme 16) [18].
![[1860-5397-7-82-i16]](/bjoc/content/inline/1860-5397-7-82-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Gold-catalyzed reactions of cyclopropenyl esters 34b–34d.
Scheme 16: Gold-catalyzed reactions of cyclopropenyl esters 34b–34d.
Interestingly, the gold-catalyzed rearrangement of cyclopropenylsilane 34e provided two compounds: Furanone 35a (40%) and indene 36a (39%) both devoid of a trimethylsilyl group. Since protodesilylation took place readily, it is likely that the allylic silanes 35’e and 36’e were the initially generated products. Their formation could be explained by regioselective electrophilic activation and ring-opening of the cyclopropenylsilane leading to the organogold 37’e (formally arising from ring-opening of a cyclopropyl cation at the β-position of the trimethylsilyl group). Subsequent nucleophilic attack by the carbonyl and the phenyl would produce 35’e and 36’e, though this was not discussed by the authors (Scheme 17) [18].
![[1860-5397-7-82-i17]](/bjoc/content/inline/1860-5397-7-82-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Gold-catalyzed reactions of cyclopropenylsilane 34e.
Scheme 17: Gold-catalyzed reactions of cyclopropenylsilane 34e.
It is worth noting that for substrates bearing two electron-withdrawing groups at C3 (COMe and CO2Me), no gold-catalyzed rearrangement took place under similar conditions. However, such cyclopropenes have been converted to furans in the presence of CuI or PdCl2(MeCN)2 as catalysts [34].
Rearrangement of cyclopropenylmethyl acetates
Propargylic carboxylates have proven to be particularly interesting substrates in gold-catalyzed reactions that have led to the development of useful synthetic processes relying on 1,3- or 1,2-acyloxy migration as the key step, depending on the substitution pattern [35,36]. Due to their high strain and π-electron density, cyclopropenes exhibit reactivity often comparable to that of alkynes in transition metal-catalyzed reactions. Not surprisingly, the reactivity of cyclopropenylmethyl carboxylates in the presence of gold catalysts has been investigated as reported in 2010 by Ariafard, Hyland et al. [22]. These authors reported that 2,3,3-trimethyl-cyclopropenylmethyl acetates 39 underwent a gold-catalyzed rearrangement into the corresponding 2-acetoxydienes 40, and a screening of gold catalysts indicated the superior activity of Gagosz’s complex [(Ph3P)AuNTf2] in terms of yield and selectivity. Starting from arylcyclopropenylmethyl acetates 39a–39e substituted by a phenyl group or an electron-deficient aromatic ring, a low temperature (CH2Cl2, −50 °C) was essential to obtain the 2-acetoxydienes 40a–40e with high Z-selectivity (Z/E = 10:1–41:1). Cyclopropenylmethyl acetate 39f substituted by the electron-rich p-tolyl group effectively underwent rearrangement, but the corresponding diene 40f decomposed rapidly. A low selectivity (Z:E = 1.8:1) was observed for the 2-acetoxydiene 40g resulting from the rearrangement of cyclopropenylmethyl acetate 39g substituted by an n-alkyl group (Scheme 18) [22].
![[1860-5397-7-82-i18]](/bjoc/content/inline/1860-5397-7-82-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Gold-catalyzed rearrangement of cyclopropenylmethyl acetates.
Scheme 18: Gold-catalyzed rearrangement of cyclopropenylmethyl acetates.
Among the conceivable mechanisms, DFT calculations indicated that the kinetically favored pathway involved an initial regioselective electrophilic activation of the cyclopropene followed by ring-opening to yield the gold-stabilized allylic carbocation 41. Subsequent 1,2-migration of the acetoxy group proceeded via the formation of five-membered intermediates 42 or 42', which then collapsed to the geometric isomers of the corresponding 2-acetoxydiene. For steric reasons, the energy barrier was found to be significantly lower for the pathway leading to the Z isomer, with a larger calculated difference when a phenyl group was present (R = Ph, 5.7 kcal·mol−1) compared to an n-alkyl substituent (R = Et, 1.6 kcal·mol−1), which correlates well with the experimental results (Scheme 19) [22].
![[1860-5397-7-82-i19]](/bjoc/content/inline/1860-5397-7-82-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Mechanism of the gold-catalyzed rearrangement of cyclopropenes 39.
Scheme 19: Mechanism of the gold-catalyzed rearrangement of cyclopropenes 39.
The gold-catalyzed reactions involving cyclopropenes examined so far in this review have involved capture of the organogold intermediates, resulting from electrophilic activation and ring-opening, by an external or an internal nucleophile. Cyclopropanation of olefins, a reaction classically attributed to the carbene-like reactivity, will now be examined.
Cyclopropanation of olefins with gold carbenes generated from cyclopropenes
Intermolecular cyclopropanation of olefins
In 2008, Lee et al. disclosed several representative gold-catalyzed reactions with cyclopropenes and reported one example of cyclopropanation achieved via the gold-carbene intermediate. Thus, when 3-methyl-3-nonylcyclopropene (8) was treated with a catalytic amount of [(Ph3P)AuNTf2] in the presence of excess styrene, the alkenyl cyclopropane 44, resulting from intermolecular cyclopropanation triggered by the gold carbene 43, was isolated in 72% yield as a 6:1 mixture of cis/trans diastereomers and a 1.6:1 mixture of Z/E-geometric isomers (Scheme 20) [18].
![[1860-5397-7-82-i20]](/bjoc/content/inline/1860-5397-7-82-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Gold-catalyzed cyclopropanation of styrene with cyclopropene 8.
Scheme 20: Gold-catalyzed cyclopropanation of styrene with cyclopropene 8.
Angelici, Woo, et al. reported several catalytic reactions of carbene precursors on bulk gold metal powder consisting of particles (5–50 µm size) prepared by reduction of HAuCl4 with hydroquinone [23]. Upon treatment with this gold powder (MeCN, 60 °C), 3,3-diphenylcyclopropene (1) gave 1,1,6,6-tetraphenylhexa-1,3,5-triene (45), arising from self-coupling of a surface bound gold carbene, as a 40:60 mixture of Z/E-geometric isomers (82%). Cross-couplings of carbenes derived from cyclopropene 1 and phenyldiazomethane or ethyl diazoacetate on bulk gold powder were also studied, but mixtures of self- and cross-coupling products were invariably obtained with negligible selectivity. Interestingly, the authors investigated the intermolecular cyclopropanation of styrene by the surface bound gold carbene generated from cyclopropene 1. Though a large excess of styrene (100 equiv) was used, triene 45 resulting from the self-coupling of 1 still predominated, and the cyclopropanation product 46 was isolated in low yield (19%) as a single trans diastereomer (Scheme 21) [23].
![[1860-5397-7-82-i21]](/bjoc/content/inline/1860-5397-7-82-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Representative reactions of carbene precursors on gold metal.
Scheme 21: Representative reactions of carbene precursors on gold metal.
In their investigations on the bonding model for gold(I) carbenoid complexes, Toste et al. highlighted the importance of the substitution pattern and the ligands (Scheme 4). Interestingly, DFT calculations were carried out for organogold species that can actually be generated by ring-opening of cyclopropenes, and therefore the authors examined experimentally the impact of cationic versus carbene-like species on the reactivity in olefin cyclopropanation [17]. In the presence of an olefin and a cationic gold(I) catalyst, cyclopropenone acetal 3 did not provide any cyclopropanation product, which is in agreement with the fact that the organogold species generated by ring-opening of 3 should instead react as a gold-stabilized carbocation due to the presence of oxygen atoms that can stabilize the cationic intermediate. However, it is worth pointing out that Boger and Brotherton previously reported that cyclopropenone acetals could cyclopropanate electron-deficient olefins, via charged intermediates, under simple thermal conditions [37]. In contrast to the behaviour of cyclopropenone acetal 3, Toste et al. observed that the reaction of the 3,3-disubstituted cyclopropene 47 and (Z)-stilbene in the presence of a cationic gold catalyst could effectively provide the desired cyclopropanation product 48, but the yield and the diastereoselectivity were highly dependent on the gold ligand. As anticipated from the structural studies, π-acidic phosphites that increase cation-like reactivity gave little or none of the cyclopropanation product 48. Phosphines gave moderate results, whereas the highest yield and diastereoselectivity was obtained when the strong σ donor and weak π acceptor N-heterocyclic carbene IPr was the ligand. The latter was indeed anticipated to give an organogold with a higher carbene-like reactivity which favors olefin cyclopropanation. AuCl was unreactive under these conditions (Scheme 22) [17].
![[1860-5397-7-82-i22]](/bjoc/content/inline/1860-5397-7-82-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Intermolecular olefin cyclopropanation with gold carbenes generated from cyclopropenes.
Scheme 22: Intermolecular olefin cyclopropanation with gold carbenes generated from cyclopropenes.
Intermolecular cyclopropanation of furans: Synthesis of conjugated trienes
In 2011, Lee and Hadfield reported the synthesis of conjugated trienes by gold-catalyzed intermolecular reaction of cyclopropenes with furans [24]. Several catalysts such as [(Ph3P)AuNTf2], or IPrAuCl in combination with different silver salts could be used successfully, but the highest yields were obtained with the cationic gold catalyst 49. In the presence of 2-methylfuran, a variety of 3,3-disubstituted cyclopropenes led to trienes 50/50’, and the initially generated mixture of geometric isomers was isomerized by treatment with a catalytic amount of iodine. Trienes 50/50’ were isolated in good yields and with satisfactory levels of stereoselectivity when the steric bulk of the two substituents (R and R’) were significantly different or if a phenyl group was present. Although no cyclopropane derivative was obtained from this reaction, this transformation has been included in this section because one of the possible mechanisms involves an initial cyclopropanation of the less hindered olefin in 2-methylfuran by the organogold intermediate 51, followed by ring-opening. The alternative mechanism involves nucleophilic attack of 2-methylfuran on the gold carbene 51, followed by elimination (Scheme 23) [24].
![[1860-5397-7-82-i23]](/bjoc/content/inline/1860-5397-7-82-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Gold-catalyzed formation of trienes from cyclopropenes and furans.
Scheme 23: Gold-catalyzed formation of trienes from cyclopropenes and furans.
The reaction was more difficult to carry out with cyclopropene carboxylates 52 and 53 possessing tetrasubstituted alkene structures. The former substrate required harsher conditions (DCE, 80 °C), but the corresponding tetrasubstituted triene 54 was still obtained in good yield (77%). For the latter substrate, the reaction was conducted in an excess of 2-methylfuran and triene 55 was isolated in low yield (37%), accompanied by dienoate 56 as a by-product (Scheme 24) [24].
![[1860-5397-7-82-i24]](/bjoc/content/inline/1860-5397-7-82-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Gold-catalyzed formation of trienes from cyclopropenes and furans.
Scheme 24: Gold-catalyzed formation of trienes from cyclopropenes and furans.
Other mono- or disubstituted furans can be successfully used as partners, as illustrated by the gold-catalyzed reactions involving 3-tert-butyl-3-methylcyclopropene as substrate that led to the corresponding tetra- or pentasubstituted trienes of type 57 or triene 58 bearing two geminal electron-deficient groups (Scheme 25) [24].
![[1860-5397-7-82-i25]](/bjoc/content/inline/1860-5397-7-82-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Gold-catalyzed formation of trienes from cyclopropenes and furans.
Scheme 25: Gold-catalyzed formation of trienes from cyclopropenes and furans.
Besides these examples of intermolecular cyclopropanations, examples of intramolecular cyclopropanation of olefins by gold carbenes generated from cyclopropenes have been investigated in our group.
Intramolecular cyclopropanation: cycloisomerization of cyclopropene-enes
In 1981, Padwa et al. reported that 1,2-diphenylcyclopropenes, substituted by allyl, methallyl, crotyl groups at C3, rearranged to the corresponding 1,2-diphenylbicyclo[3.1.0]hex-2-enes upon treatment with nearly stoichiometric quantities of AgClO4 and prolonged heating in C6H6 or MeOH at reflux [33,38]. These reactions appear to constitute the first examples of intramolecular olefin cyclopropanation promoted by a silver carbenoid generated by ring-opening of a cyclopropene.
We envisioned that the gold carbene resulting from the ring-opening of appropriately substituted cyclopropenes could also be involved in intramolecular olefin cyclopropanation in order to access [n.1.0] bicyclic ring systems. Rather than examining the behaviour of cyclopropenes bearing an allylic chain at C3, and in order to avoid aryl-substituted cyclopropenes that have routinely been used as substrates, allylic ethers derived from cyclopropenyl carbinols were selected as substrates. Cyclopropenyl carbinols have recently emerged as synthetically useful building blocks [39] and are readily available by the condensation of an in situ generated cyclopropenyl organolithium with an aldehyde [39,40]. Additionally, they can be obtained in an enantiomerically enriched form by Sharpless kinetic resolution [41]. In order to ensure regioselective ring-opening of the cyclopropene ring, cyclopropenyl carbinols possessing a trisubstituted endocyclic alkene were considered with the hope that a secondary cyclopropyl cation would be preferentially formed upon coordination of a gold complex. However, this implies that substituents have to be present at C3 in order to handle stable substrates. Thus, allyl 3,3-dimethylcyclopropenylcarbinyl ether 59 was prepared and several gold(I) and gold(III) species {AuCl3, AuBr3, AuCl, [(Ph3P)AuNTf2], [(Ph3P)AuSbF6] or [(Ph3P)AuOTf]} were found to catalyze smoothly the cycloisomerization and yield the desired oxabicyclic compound 60 in high yields and with excellent diastereoselectivity (dr > 96:4) [25]. The observed stereochemical outcome has been tentatively rationalized by considering a twist-boat transition state model in which the gold center and the allylic benzyloxymethyl substituents both occupy axial positions in order to avoid 1,3-allylic strain with the vinylic methyl groups. The isopropylidene group in compound 60 can be cleaved by ozonolysis to give the corresponding 3-oxabicyclo[4.1.0]heptanone 61 (85%), and hence, the 3,3-dimethylcyclopropene moiety appears to be an excellent surrogate of an α-diazoketone (Scheme 26) [25,42].
![[1860-5397-7-82-i26]](/bjoc/content/inline/1860-5397-7-82-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Gold-catalyzed cycloisomerization of cyclopropene-ene 59.
Scheme 26: Gold-catalyzed cycloisomerization of cyclopropene-ene 59.
Further studies were carried out using AuCl as a catalyst and the reaction was generalized for a variety of substituted allylic ethers 62a–62f. Excellent results were obtained with allylic ethers bearing one (62a, 62b) or two substituents (62c–62e) at the terminal position of the olefin and the corresponding oxabicyclic compounds were isolated in high yields (93–99%). The stereospecificity of the cyclopropanation process was highlighted by the behaviour of geranyl ether 62d and neryl ether 62e, which furnished the epimeric cycloisomerization products 63d and 63e, respectively. The stereoselectivity was lower for methallyl ether 62f which afforded compound 63f as an 87:13 mixture of diastereomers (Scheme 27) [25].
![[1860-5397-7-82-i27]](/bjoc/content/inline/1860-5397-7-82-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Gold-catalyzed cycloisomerization of substituted allyl cyclopropenyl carbinyl ethers 62a–62f.
Scheme 27: Gold-catalyzed cycloisomerization of substituted allyl cyclopropenyl carbinyl ethers 62a–62f.
The influence of the substituent at the α-position of the oxygen atom and the cyclopropene has also been examined. Diastereoselectivities and yields were always high when this substituent was branched, whatever the relative configuration of the additional stereocenter, as shown with substrates 64a–64e and 66 which led to the oxabicyclic products 65a–65e and 67, respectively. The substituent could also be a longer linear n-alkyl chain functionalized at the remote position by a benzyl ether, as illustrated for the cycloisomerization of 68 to 69. Interestingly, the azabicyclic compound 71 was obtained in excellent yield (99%) and with high diastereoselectivity (dr > 96:4) by gold-catalyzed cycloisomerization of the N-allyl sulfonamide 70 (Scheme 28) [25].
![[1860-5397-7-82-i28]](/bjoc/content/inline/1860-5397-7-82-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Gold-catalyzed cycloisomerization of cyclopropene-enes.
Scheme 28: Gold-catalyzed cycloisomerization of cyclopropene-enes.
The success of the gold-catalyzed cycloisomerization of cyclopropene-enes, proceeding with intramolecular cyclopropanation of the olefin, lies in the chemoselective activation of the cyclopropene, in preference to the alkene, which allows the generation of a gold carbene intermediate. The relative reactivity of cyclopropenes compared to alkynes is an interesting issue that has been addressed by Wang et al. during their studies on the gold-catalyzed cycloisomerization of cyclopropene-ynes [26].
Cycloisomerization of cyclopropene-ynes
Upon treatment with [(Ph3P)AuOTf] (5 mol %), several propargylic alcohols possessing a 2,3-diphenylcycloprop-2-enyl substituent were smoothly converted (CH2Cl2, rt, 5 min) to substituted 4,5-diphenylphenols. The scope of the reaction is quite broad since it could be applied to secondary propargylic alcohols 72 (or an acetate derivative 73), to tertiary alcohols such as 74 or 75 and even to the O-trimethylsilyl cyanohydrin 76. The corresponding cycloisomerization products 77–81 were isolated in good to excellent yields (71–97%). The hydroxyl group did not exert a particular role in this process since 1,2-diphenyl-3-propargylcyclopropene was rearranged to 1,2-diphenylbenzene (97%) under the same conditions [26]. The formation of phenols (and their derivatives) 77–81 could be explained by an initial chemoselective activation of the alkyne by the gold catalyst with subsequent intramolecular nucleophlic attack of the cyclopropene olefin. Ring-opening of the cyclopropyl cation E to generate the 1,3-cyclohexadiene F and a 1,2-shift of the R1 group would then lead to the substituted 4,5-diphenylphenol. In this mechanistic pathway, the cyclopropene carbon atoms become directly linked to those of the alkyne with no skeletal rearrangement (Scheme 29) [26].
![[1860-5397-7-82-i29]](/bjoc/content/inline/1860-5397-7-82-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Gold-catalyzed cycloisomerization of cyclopropene-ynes.
Scheme 29: Gold-catalyzed cycloisomerization of cyclopropene-ynes.
However, the substituents were found to exert an important influence on the outcome of the reaction. Indeed, the secondary propargylic alcohols 82a and 82b, in which the alkyne is terminal or substituted by an n-pentyl group, afforded an equimolar mixture of two regioisomeric phenols (91–99%). Whereas 4,5-diphenylphenols 83a and 83b correspond to the previously observed rearrangement pathway, the structure of the symmetrical phenols 84a and 84b indicates that cleavage of both the cyclopropene double bond and the alkyne had occurred. To explain the formation of the latter double cleavage products, Wang et al. proposed a mechanistic scenario in which back donation from gold in the initially formed vinyl gold species E led to the highly strained gold carbene G possessing a tricyclo[3.1.0.02,6]hexane structure. Rearrangement of G by consecutive 1,2-alkyl shifts, proceeding through carbocations H and I and Dewar-type benzene J as intermediates, followed by ring-opening and a 1,2-hydrogen shift, ultimately led to 84a or 84b (Scheme 30) [26].
![[1860-5397-7-82-i30]](/bjoc/content/inline/1860-5397-7-82-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Formation of products arising from a double cleavage process in the gold-catalyzed cycloisomerization of cyclopropene-ynes.
Scheme 30: Formation of products arising from a double cleavage process in the gold-catalyzed cycloisomerizati...
Since intermediates G–J are all sterically crowded, this double cleavage mechanistic pathway should be favored for cyclopropenes bearing smaller substituents. In fact, for cyclopropenes 85a–85c having two n-butyl substituents or cyclopropenes 86a–86d with one n-butyl and one trimethylsilyl group (the latter ensuring regioselective attack of the cyclopropene onto the activated alkyne to form a β-silylcyclopropyl cation), the gold-catalyzed rearrangement led exclusively to the phenols 87a–87c and 88a–88d (84–93%), respectively, resulting from a double cleavage process, whatever the substituent on the alkyne (Scheme 31) [26].
![[1860-5397-7-82-i31]](/bjoc/content/inline/1860-5397-7-82-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Gold-catalyzed cycloisomerization of cyclopropene-ynes involving a double cleavage process.
Scheme 31: Gold-catalyzed cycloisomerization of cyclopropene-ynes involving a double cleavage process.
Wang et al. also examined the behaviour of other cyclopropen-1,n-ynes. For substrates 89a and 89b possessing a 1,6-enyne moiety, the gold-catalyzed cycloisomerization led to the tricyclic hydrocarbons 90a (80%) and 90b (74%), respectively. The alkyne, chemoselectively activated by the gold complex, underwent nucleophilic attack by the cyclopropene in a 5-exo-dig manner followed by ring-opening. A subsequent Friedel–Crafts cyclization allowed the formation of the indene subunit (Equation 1, Scheme 32). Sulfonamide 91 contains a 1,7-enyne subunit and its gold-catalyzed cycloisomerization delivered tricyclic compound 92 incorporating a seven-membered nitrogen heterocycle. The yield of this transformation was found to be greatly improved when in situ generated [(JohnPhos)AuSbF6] was used as the catalyst (88%) instead of [(Ph3P)AuOTf] (30%) (Equation 2, Scheme 32). When the alkyne was replaced by an alkene or an allene, the corresponding substrates 93 and 94 underwent a gold-catalyzed rearrangement to afford indenes 95 (80%) and 96 (78%), respectively. Interestingly, only the cyclopropene reacted by ring-opening followed by Friedel–Crafts cyclization: The alkene and the allene units were unaffected (Equation 3, Scheme 32) [26].
![[1860-5397-7-82-i32]](/bjoc/content/inline/1860-5397-7-82-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Gold-catalyzed reaction of cyclopropene-ynes, cyclopropene-enes and cyclopropene-allenes.
Scheme 32: Gold-catalyzed reaction of cyclopropene-ynes, cyclopropene-enes and cyclopropene-allenes.
Thus, alkynes appear to be chemoselectively activated in the presence of gold complexes in preference to cyclopropenes, whereas the latter moiety is more reactive than alkenes and possibly allenes, although in the latter case only a single example of competition was reported.
Conclusion
Though relatively recent, the entry of cyclopropenes into the area of gold catalysis has already led to interesting contributions exploiting different aspects of the reactivity of alkenyl organogold carbenoids. It is obvious that the possibility to generate gold carbenes from cyclopropenes opens new possibilities and further synthetic developments in this field will certainly be reported.
References
-
Hoffmann-Röder, A.; Krause, N. Org. Biomol. Chem. 2005, 3, 387–391. doi:10.1039/b416516k
Return to citation in text: [1] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081
Return to citation in text: [1] -
Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082
Return to citation in text: [1] -
Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018
Return to citation in text: [1] -
Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j
Return to citation in text: [1] -
Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589
Return to citation in text: [1] -
Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l
Return to citation in text: [1] -
Seidel, G.; Mynott, R.; Fürstner, A. Angew. Chem., Int. Ed. 2009, 48, 2510–2513. doi:10.1002/anie.200806059
Return to citation in text: [1] [2] [3] -
Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331
Return to citation in text: [1] [2] [3] [4] [5] -
Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370
Return to citation in text: [1] [2] [3] [4] [5] -
Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Seraya, E.; Slack, E.; Ariafard, A.; Yates, B. F.; Hyland, C. J. T. Org. Lett. 2010, 12, 4768–4771. doi:10.1021/ol101862u
Return to citation in text: [1] [2] [3] [4] -
Zhou, Y.; Trewyn, B. G.; Angelici, R. J.; Woo, L. K. J. Am. Chem. Soc. 2009, 131, 11734–11743. doi:10.1021/ja900653s
Return to citation in text: [1] [2] [3] -
Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j
Return to citation in text: [1] [2] [3] [4] [5] -
Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f
Return to citation in text: [1] [2] [3] [4] [5] -
Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Fürstner, A.; Morency, L. Angew. Chem., Int. Ed. 2008, 47, 5030–5033. doi:10.1002/anie.200800934
Return to citation in text: [1] -
Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917
Return to citation in text: [1] -
Hadfield, M. S.; Lee, A.-L. Org. Lett. 2010, 12, 484–487. doi:10.1021/ol902675k
Return to citation in text: [1] [2] -
Witham, C. A.; Mauléon, P.; Shapiro, N. D.; Sherry, B. D.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 5838–5839. doi:10.1021/ja071231+
Return to citation in text: [1] -
Shao, L.-X.; Zhang, Y.-P.; Qi, M.-H.; Shi, M. Org. Lett. 2007, 9, 117–120. doi:10.1021/ol0626746
Return to citation in text: [1] [2] -
Müller, P.; Pautex, N.; Doyle, M. P.; Bagheri, V. Helv. Chim. Acta 1990, 73, 1233–1241. doi:10.1002/hlca.19900730513
Return to citation in text: [1] -
Padwa, A.; Blacklock, T.; Loza, R. J. Am. Chem. Soc. 1981, 103, 2404–2405. doi:10.1021/ja00399a042
Return to citation in text: [1] [2] -
Ma, S.; Zhang, J. J. Am. Chem. Soc. 2003, 125, 12386–12387. doi:10.1021/ja036616g
Return to citation in text: [1] -
Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773
Return to citation in text: [1] -
Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769
Return to citation in text: [1] -
Boger, D. L.; Brotherton, C. E. Tetrahedron Lett. 1984, 25, 5611–5614. doi:10.1016/S0040-4039(01)91393-0
Return to citation in text: [1] -
Padwa, A.; Blacklock, T. J.; Loza, R. J. Org. Chem. 1982, 47, 3712–3721. doi:10.1021/jo00140a025
Return to citation in text: [1] -
Simaan, S.; Masarwa, A.; Zohar, E.; Stanger, A.; Bertus, P.; Marek, I. Chem.–Eur. J. 2009, 15, 8449–8464. doi:10.1002/chem.200901074
Return to citation in text: [1] [2] -
Baird, M. S.; Hussain, H. H.; Nethercott, W. J. Chem. Soc., Perkin Trans. 1 1986, 1845–1853. doi:10.1039/P19860001845
Return to citation in text: [1] -
Simaan, S.; Masarwa, A.; Bertus, P.; Marek, I. Angew. Chem., Int. Ed. 2006, 45, 3963–3965. doi:10.1002/anie.200600556
Return to citation in text: [1] -
Hon, Y.-S.; Chang, R.-C. Heterocycles 2008, 32, 1089–1099. doi:10.3987/COM-90-5626
Return to citation in text: [1]
| 32. | Müller, P.; Pautex, N.; Doyle, M. P.; Bagheri, V. Helv. Chim. Acta 1990, 73, 1233–1241. doi:10.1002/hlca.19900730513 |
| 33. | Padwa, A.; Blacklock, T.; Loza, R. J. Am. Chem. Soc. 1981, 103, 2404–2405. doi:10.1021/ja00399a042 |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 35. | Marion, N.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2750–2752. doi:10.1002/anie.200604773 |
| 36. | Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Angew. Chem., Int. Ed. 2008, 47, 718–721. doi:10.1002/anie.200703769 |
| 22. | Seraya, E.; Slack, E.; Ariafard, A.; Yates, B. F.; Hyland, C. J. T. Org. Lett. 2010, 12, 4768–4771. doi:10.1021/ol101862u |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 34. | Ma, S.; Zhang, J. J. Am. Chem. Soc. 2003, 125, 12386–12387. doi:10.1021/ja036616g |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 22. | Seraya, E.; Slack, E.; Ariafard, A.; Yates, B. F.; Hyland, C. J. T. Org. Lett. 2010, 12, 4768–4771. doi:10.1021/ol101862u |
| 22. | Seraya, E.; Slack, E.; Ariafard, A.; Yates, B. F.; Hyland, C. J. T. Org. Lett. 2010, 12, 4768–4771. doi:10.1021/ol101862u |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 24. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 24. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 17. | Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331 |
| 24. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 17. | Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331 |
| 37. | Boger, D. L.; Brotherton, C. E. Tetrahedron Lett. 1984, 25, 5611–5614. doi:10.1016/S0040-4039(01)91393-0 |
| 23. | Zhou, Y.; Trewyn, B. G.; Angelici, R. J.; Woo, L. K. J. Am. Chem. Soc. 2009, 131, 11734–11743. doi:10.1021/ja900653s |
| 23. | Zhou, Y.; Trewyn, B. G.; Angelici, R. J.; Woo, L. K. J. Am. Chem. Soc. 2009, 131, 11734–11743. doi:10.1021/ja900653s |
| 33. | Padwa, A.; Blacklock, T.; Loza, R. J. Am. Chem. Soc. 1981, 103, 2404–2405. doi:10.1021/ja00399a042 |
| 38. | Padwa, A.; Blacklock, T. J.; Loza, R. J. Org. Chem. 1982, 47, 3712–3721. doi:10.1021/jo00140a025 |
| 39. | Simaan, S.; Masarwa, A.; Zohar, E.; Stanger, A.; Bertus, P.; Marek, I. Chem.–Eur. J. 2009, 15, 8449–8464. doi:10.1002/chem.200901074 |
| 24. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 1. | Hoffmann-Röder, A.; Krause, N. Org. Biomol. Chem. 2005, 3, 387–391. doi:10.1039/b416516k |
| 2. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368 |
| 3. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 4. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 5. | Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395–403. doi:10.1038/nature05592 |
| 6. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 7. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 8. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 9. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 10. | Shen, H. C. Tetrahedron 2008, 64, 3885–3903. doi:10.1016/j.tet.2008.01.081 |
| 11. | Shen, H. C. Tetrahedron 2008, 64, 7847–7870. doi:10.1016/j.tet.2008.05.082 |
| 12. | Muzart, J. Tetrahedron 2008, 64, 5815–5849. doi:10.1016/j.tet.2008.04.018 |
| 13. | Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208–3221. doi:10.1039/b816696j |
| 14. | Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. doi:10.1002/anie.200701589 |
| 16. | Seidel, G.; Mynott, R.; Fürstner, A. Angew. Chem., Int. Ed. 2009, 48, 2510–2513. doi:10.1002/anie.200806059 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 27. | Fürstner, A.; Morency, L. Angew. Chem., Int. Ed. 2008, 47, 5030–5033. doi:10.1002/anie.200800934 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 16. | Seidel, G.; Mynott, R.; Fürstner, A. Angew. Chem., Int. Ed. 2009, 48, 2510–2513. doi:10.1002/anie.200806059 |
| 17. | Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 20. | Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 22. | Seraya, E.; Slack, E.; Ariafard, A.; Yates, B. F.; Hyland, C. J. T. Org. Lett. 2010, 12, 4768–4771. doi:10.1021/ol101862u |
| 23. | Zhou, Y.; Trewyn, B. G.; Angelici, R. J.; Woo, L. K. J. Am. Chem. Soc. 2009, 131, 11734–11743. doi:10.1021/ja900653s |
| 24. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 25. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 25. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 15. | Rubin, M.; Rubina, M.; Gevorgyan, V. Chem. Rev. 2007, 107, 3117–3179. doi:10.1021/cr050988l |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 25. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 17. | Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 25. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 17. | Benitez, D. N.; Shapiro, N. D.; Tkatchouk, E.; Wang, Y.; Goddard, W. A., III; Toste, F. D. Nat. Chem. 2009, 1, 482–486. doi:10.1038/nchem.331 |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 25. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 42. | Hon, Y.-S.; Chang, R.-C. Heterocycles 2008, 32, 1089–1099. doi:10.3987/COM-90-5626 |
| 16. | Seidel, G.; Mynott, R.; Fürstner, A. Angew. Chem., Int. Ed. 2009, 48, 2510–2513. doi:10.1002/anie.200806059 |
| 39. | Simaan, S.; Masarwa, A.; Zohar, E.; Stanger, A.; Bertus, P.; Marek, I. Chem.–Eur. J. 2009, 15, 8449–8464. doi:10.1002/chem.200901074 |
| 40. | Baird, M. S.; Hussain, H. H.; Nethercott, W. J. Chem. Soc., Perkin Trans. 1 1986, 1845–1853. doi:10.1039/P19860001845 |
| 28. | Mézailles, N.; Ricard, L.; Gagosz, F. Org. Lett. 2005, 7, 4133–4136. doi:10.1021/ol0515917 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 41. | Simaan, S.; Masarwa, A.; Bertus, P.; Marek, I. Angew. Chem., Int. Ed. 2006, 45, 3963–3965. doi:10.1002/anie.200600556 |
| 30. | Witham, C. A.; Mauléon, P.; Shapiro, N. D.; Sherry, B. D.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 5838–5839. doi:10.1021/ja071231+ |
| 29. | Hadfield, M. S.; Lee, A.-L. Org. Lett. 2010, 12, 484–487. doi:10.1021/ol902675k |
| 29. | Hadfield, M. S.; Lee, A.-L. Org. Lett. 2010, 12, 484–487. doi:10.1021/ol902675k |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 21. | Li, C.; Zeng, Y.; Wang, J. Tetrahedron Lett. 2009, 50, 2956–2959. doi:10.1016/j.tetlet.2009.03.203 |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 20. | Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 31. | Shao, L.-X.; Zhang, Y.-P.; Qi, M.-H.; Shi, M. Org. Lett. 2007, 9, 117–120. doi:10.1021/ol0626746 |
| 20. | Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 31. | Shao, L.-X.; Zhang, Y.-P.; Qi, M.-H.; Shi, M. Org. Lett. 2007, 9, 117–120. doi:10.1021/ol0626746 |
| 20. | Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
| 18. | Bauer, J. T.; Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2008, 6405–6407. doi:10.1039/b815891f |
| 19. | Hadfield, M. S.; Bauer, J. T.; Glen, P. E.; Lee, A.-L. Org. Biomol. Chem. 2010, 8, 4090–4095. doi:10.1039/C0OB00085J |
| 26. | Li, C.; Zeng, Y.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 6413–6417. doi:10.1002/anie.201002673 |
| 20. | Zhu, Z. B.; Shi, M. Chem.–Eur. J. 2008, 14, 10219–10222. doi:10.1002/chem.200801370 |
© 2011 Miege et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)