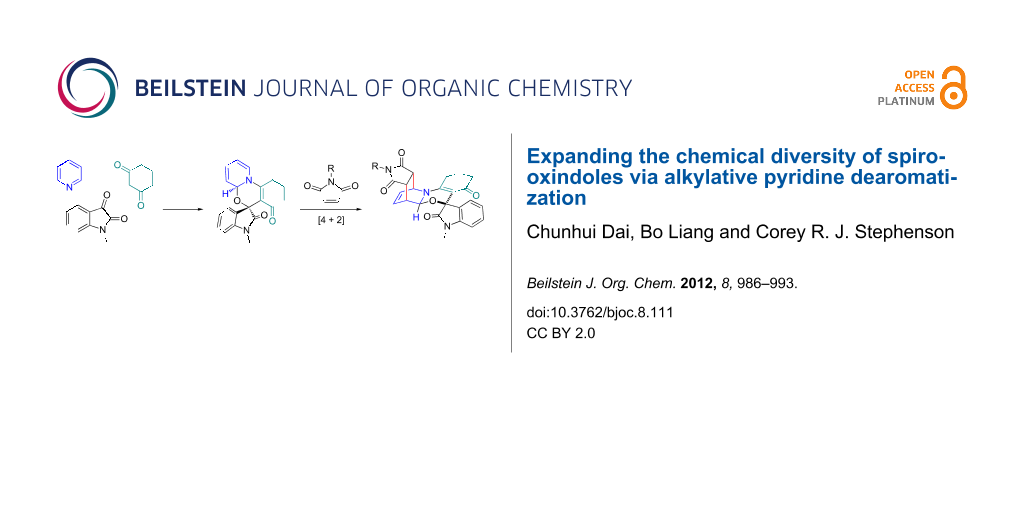
Abstract
A mild and practical synthesis of spirooxindole [1,3]oxazino derivatives from N-substituted isatins and 1,3-dicarbonyl compounds with pyridine derivatives is reported. The reactions provided good to excellent yields. Further exploration of the molecular diversity of these compounds is demonstrated through Diels–Alder reactions.

Graphical Abstract
Introduction
The spirooxindole is a common structural motif found in a variety of complex alkaloids [1]. Many compounds that possess a spirooxindole moiety exhibit significant biological activity, thus exemplifying their role in drug development [2-8]. Moreover, the challenging molecular architecture of spirooxindoles is appealing to chemists because it evokes novel synthetic strategies that address configurational demands and provides platforms for further reaction development [9-11]. To our knowledge, most studies of these types of molecules focus on spirooxindoles bearing a pyrrolidine ring at the 3-position of the oxindole core, while few reports expand to formulate the syntheses of other spiro rings. As part of our ongoing reaction-screening objective [12,13], we previously reported a Lewis acid catalyzed, three-component synthesis of spirooxindole pyranochromenedione derivatives using isatin and two 1,3-dicarbonyl compounds (Scheme 1) [14]. Mechanistically, we believed this reaction to proceed through an intermediate isatylidene 1 [15-17]. As a means to support our mechanistic hypothesis, we attempted to prepare and isolate the isatylidene; however, our attempts were unsuccessful. Interestingly, treatment of 2 with methanesulfonyl chloride (MsCl) in pyridine provided the dearomatized alkylation product 3 in 78% yield. In the context of developing novel and practical methods for the preparation of diverse heterocyclic compounds, herein, we report our extended investigation on the efficient synthesis of spirooxindole [1,3]oxazino derivatives by means of alkylative pyridine dearomatization [18-21].
![[1860-5397-8-111-i1]](/bjoc/content/inline/1860-5397-8-111-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Unexpected alkylative pyridine dearomatization during our previous work on the synthesis of spirooxindole pyranochromenediones.
Scheme 1: Unexpected alkylative pyridine dearomatization during our previous work on the synthesis of spiroox...
Results and Discussion
Reactions of N-substituted isatins and 1,3-dicarbonyl compounds in pyridine
Based on preliminary results, we commenced with the reaction of N-substituted isatins 4 and 1,3-dicarbonyl compounds 5 in pyridine. After addition of the reagents, the mixture was allowed to react at room temperature for 2 h to ensure initial coupling, whereupon methanesulfonyl chloride was added slowly over a 1 h period at 0 °C and another 2 h at the same temperature to trap the vinylogous acid as vinyl mesylates 6. Various N-substituted isatins and 1,3-dicarbonyl compounds were then explored and the results are presented in Table 1. Beginning with isatin and 1,3-cyclohexanedione (5a) as coupling partners, we isolated a relatively poor yield of product 6a (Table 1, entry 1). We speculated that the free indole nitrogen was inhibiting the reaction, thus we switched to N-substituted isatins and found that the reaction improved to provide moderate to high yields (Table 1, entries 2–10). Subtle substitution effects were observed when the C(5)–H of isatin was replaced with various functionalities. Specifically, an electron-donating group at the 5-position, such as a methyl group, decreased the reactivity and only gave 57% yield (Table 1, entry 3). As a comparison, electron-withdrawing groups at the 5-position, such as chloro and nitro groups, increased the reactivity and provided products in higher yield (Table 1, entries 4–5). Other 1,3-dicarbonyl compounds were also investigated. 5,5-Dimethyl-1,3-cyclohexanedione (5b) worked well (Table 1, entry 6), in contrast to 5-phenyl-1,3-cyclohexanedione (5c), which gave a slightly lower yield (Table 1, entry 7). Interestingly, nonequivalent 1,3-dicarbonyl compound 5d afforded single constitutional isomer (Table 1, entry 8), presumably due to the increased sterics and overall ring strain associated with substituents alpha to the vinylogous sulfonyl ester. N-Phenylisatin provided a similar yield to N-methylisatin (Table 1, entry 9). However, when N-acetylisatin was subjected to the reaction conditions, the reaction failed to provide the desired product and instead delivered compound 6j exclusively in 74% yield (Table 1, entry 10). We predict that the formation of 6j by dehydration is due to the electron deficiency of the oxindole ring and subsequent stability gained from the ene–trione moiety. The structure of compound 6b was established by single-crystal X-ray analysis (Figure 1).
Table 1: Reaction of isatins 4 and 1,3-dicarbonyl compounds 5 in pyridine.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-111-i3.svg?max-width=637&scale=1.0)
|
|||||
| entrya | R1 | R2 | 5 | product |
yield
(%)b |
|---|---|---|---|---|---|
| 1 | H | H |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-111-i4.svg?max-width=637&scale=1.0)
5a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-111-i5.svg?max-width=637&scale=1.0)
6a |
31 |
| 2 | Me | H | 5a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-111-i6.svg?max-width=637&scale=1.0)
6b |
80 |
| 3 | Me | 5-Me | 5a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-111-i7.svg?max-width=637&scale=1.0)
6c |
57 |
| 4 | Me | 5-Cl | 5a |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-111-i8.svg?max-width=637&scale=1.0)
6d |
84 |
| 5 | Me | 5-NO2 | 5a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-111-i9.svg?max-width=637&scale=1.0)
6e |
89 |
| 6 | Me | H |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-111-i10.svg?max-width=637&scale=1.0)
5b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-111-i11.svg?max-width=637&scale=1.0)
6f |
87 |
| 7 | Me | H |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-111-i12.svg?max-width=637&scale=1.0)
5c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-111-i13.svg?max-width=637&scale=1.0)
6g |
61 |
| 8 | Me | H |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-111-i14.svg?max-width=637&scale=1.0)
5d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-111-i15.svg?max-width=637&scale=1.0)
6h |
71 |
| 9 | Ph | H | 5a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-111-i16.svg?max-width=637&scale=1.0)
6i |
73 |
| 10c | Ac | H | 5a |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-111-i17.svg?max-width=637&scale=1.0)
6j |
74 |
aReactions were carried out on a 10 mmol scale in pyridine (8.0 mL) with 1.0 equiv of isatins 4 and 1,3-dicarbonyl compounds 5 at room temperature for 2 h, followed by the addition of 1.5 equiv of MsCl at 0 °C over 1 h and another 2 h stirring at the same temperature. bIsolated yield. cThe adduct 6j was isolated as the only product.
![[1860-5397-8-111-1]](/bjoc/content/figures/1860-5397-8-111-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of compound 6b.
Figure 1: X-ray crystal structure of compound 6b.
Having established a facile route to our desired vinylogous sulfonyl esters 6, we next examined their reactivity towards alkylative pyridine dearomatization reactions (Table 2). During optimization studies, we discovered that the reaction performed best at 45 °C using pyridine as the solvent. The substituent groups on the isatin moiety did not have a great effect on the reactivity of compounds 6. Generally, the reaction was complete in 12 h and provided inseparable diastereoisomers with good to excellent yields (Table 2, entries 1–5). 4-Picoline (7b) gave a lower yield after 12 h at 45 °C (Table 2, entry 6). 4-Methoxypyridine (7c) provided the desired product 3g in 78% isolated yield in only 6 h at room temperature (Table 2, entry 7). Isoquinoline (7d) and 4-acetylpyridine (7e) also underwent the reaction to provide the desired products in moderate yields (Table 2, entries 8–9), but required an elevated temperature (60 °C) and a longer reaction time (18 h). On the basis of the results in Table 2, we speculate that the unexpected formation of compound 3 in Scheme 1 was due to the evaporation of solvent after the reaction at elevated temperature (45 °C), providing an opportunity for the vinylogous sulfonate ester, generated during the reaction, to react with pyridine. Finally, the structure of compound 3d was established by single-crystal X-ray analysis (Figure 2).
Table 2: Reactions of vinylogous sulfonyl esters 6 with pyridine derivatives 7.
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-111-i18.svg?max-width=637&scale=1.0)
|
|||||||
| entrya | 6 | 7 | T (°C) | h | product 3 | drb | yield (%)c |
|---|---|---|---|---|---|---|---|
| 1 | 6b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-111-i19.svg?max-width=637&scale=1.0)
7a |
45 | 12 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-111-i20.svg?max-width=637&scale=1.0)
3a |
5:1 | 90 |
| 2 | 6c | 7a | 45 | 12 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-111-i21.svg?max-width=637&scale=1.0)
3b |
9:1 | 83 |
| 3 | 6d | 7a | 45 | 12 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-111-i22.svg?max-width=637&scale=1.0)
3c |
7:1 | 90 |
| 4 | 6e | 7a | 45 | 12 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-111-i23.svg?max-width=637&scale=1.0)
3d |
5:1 | 91 |
| 5 | 6i | 7a | 45 | 12 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-111-i24.svg?max-width=637&scale=1.0)
3e |
5:1 | 74 |
| 6 | 6b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-111-i25.svg?max-width=637&scale=1.0)
7b |
45 | 12 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-111-i26.svg?max-width=637&scale=1.0)
3f |
8:1 | 48 |
| 7 | 6b |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-111-i27.svg?max-width=637&scale=1.0)
7c |
23 | 6 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-111-i28.svg?max-width=637&scale=1.0)
3g |
3:1 | 78 |
| 8 | 6b |
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-111-i29.svg?max-width=637&scale=1.0)
7d |
60 | 18 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-111-i30.svg?max-width=637&scale=1.0)
3h |
2:1 | 65 |
| 9 | 6d |
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-111-i31.svg?max-width=637&scale=1.0)
7e |
60 | 18 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-111-i32.svg?max-width=637&scale=1.0)
3i |
8:1 | 67 |
aReactions were conducted with vinylogous sulfonyl esters 6 (1.0 mmol) and pyridine derivatives 7 (1.0 mL) at 23–60 °C. Reaction time varied from 6–18 h. bDetermined by 1H NMR integration. cIsolated yield.
![[1860-5397-8-111-2]](/bjoc/content/figures/1860-5397-8-111-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of compound 3d.
Figure 2: X-ray crystal structure of compound 3d.
Further plans to expand the molecular diversity of these compounds utilizing available functionalities are currently underway. As an illustrative example, spiro [1,3]oxazino compounds having a diene moiety within their molecular framework are susceptible to Diels–Alder (D–A) reactions [22]. Scheme 2 highlights three examples in which compound 3a was exposed to N-substituted maleimides in toluene at 150 °C under microwave irradiation for 0.5 h, and D–A products 8a–c were isolated in moderate yields. Finally, the structure of compound 8a was established by single-crystal X-ray analysis (Figure 3).
![[1860-5397-8-111-i2]](/bjoc/content/inline/1860-5397-8-111-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Application of spiro [1,3]oxazino compound 3a in D–A reactions.
Scheme 2: Application of spiro [1,3]oxazino compound 3a in D–A reactions.
![[1860-5397-8-111-3]](/bjoc/content/figures/1860-5397-8-111-3.png?scale=2.4&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of compound 8a.
Figure 3: X-ray crystal structure of compound 8a.
Conclusion
In summary, we developed a practical and efficient method to synthesize spirooxindole derivatives with a [1,3]oxazine fused-ring system. The reaction conditions are very mild and tolerant of functional groups, providing moderate to high yields. The application and versatility of these spirooxindole derivatives to quickly access complex molecules is further demonstrated in good yielding D–A reactions.
Supporting Information
| Supporting Information File 1: Full experimental details and analytical data and crystallographic information. | ||
| Format: PDF | Size: 2.6 MB | Download |
Acknowledgements
Financial support for this research from the NIH-NIGMS (P50-GM067041), NSF (CHE-1056568), the Alfred P. Sloan Foundation, Amgen, Boehringer Ingelheim, and Boston University is gratefully acknowledged. C.D. thanks AstraZeneca for a graduate fellowship. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the NSF. The authors are grateful to Professor John Porco (Boston University) for helpful discussions.
References
-
Bindra, J. S. In The Alkaloids; Manske, R. H. F., Ed.; Academic Press: New York, 1973; Vol. 14, pp 84–121.
Return to citation in text: [1] -
Onishi, T.; Sebahar, P. R.; Williams, R. M. Tetrahedron 2004, 60, 9503–9515. doi:10.1016/j.tet.2004.07.047
Return to citation in text: [1] -
Onishi, T.; Sebahar, P. R.; Williams, R. M. Org. Lett. 2003, 5, 3135–3137. doi:10.1021/ol0351910
Return to citation in text: [1] -
Sebahar, P. R.; Williams, R. M. J. Am. Chem. Soc. 2000, 122, 5666–5667. doi:10.1021/ja001133n
Return to citation in text: [1] -
Alper, P. B.; Meyers, C.; Lerchner, A.; Siegel, D. R.; Carreira, E. M. Angew. Chem., Int. Ed. 1999, 38, 3186–3189. doi:10.1002/(SICI)1521-3773(19991102)38:21<3186::AID-ANIE3186>3.0.CO;2-E
Return to citation in text: [1] -
Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N. J. Am. Chem. Soc. 1999, 121, 2147–2155. doi:10.1021/ja983788i
Return to citation in text: [1] -
Matsuura, T.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6500–6503. doi:10.1021/ja980788+
Return to citation in text: [1] -
Ashimori, A.; Bachand, B.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6477–6487. doi:10.1021/ja980786p
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 18, 3003–3025. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Beeler, A. B.; Su, S.; Singleton, C. A.; Porco, J. A., Jr. J. Am. Chem. Soc. 2007, 129, 1413–1419. doi:10.1021/ja0674744
Return to citation in text: [1] -
Balthaser, B. R.; Maloney, M. C.; Beeler, A. B.; Porco, J. A., Jr.; Snyder, J. K. Nat. Chem. 2011, 3, 969–973. doi:10.1038/nchem.1178
Return to citation in text: [1] -
Liang, B.; Kalidindi, S.; Porco, J. A., Jr.; Stephenson, C. R. J. Org. Lett. 2010, 12, 572–575. doi:10.1021/ol902764k
Return to citation in text: [1] -
Yin, X.-G.; Liu, X.-Y.; Hu, Z.-P.; Yan, M. Org. Biomol. Chem. 2012, 10, 1506–1509. doi:10.1039/C2OB06995D
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f
Return to citation in text: [1] -
Hari Babu, T.; Joseph, A. A.; Muralidharan, D.; Perumal, P. T. Tetrahedron Lett. 2010, 51, 994–996. doi:10.1016/j.tetlet.2009.12.082
Return to citation in text: [1] -
Roche, S. P.; Porco, J. A., Jr. Angew. Chem., Int. Ed. 2011, 50, 4068–4093. doi:10.1002/anie.201006017
Return to citation in text: [1] -
Nair, V.; Devipriya, S.; Suresh, E. Tetrahedron 2008, 64, 3567–3577. doi:10.1016/j.tet.2008.01.106
Return to citation in text: [1] -
Yavari, I.; Mokhtarporyani-Sanandaj, A.; Moradi, L. Tetrahedron Lett. 2007, 48, 6709–6712. doi:10.1016/j.tetlet.2007.07.135
Return to citation in text: [1] -
Nair, V.; Sreekanth, A. R.; Abhilash, N.; Biju, A. T.; Devi, B. R.; Menon, R. S.; Rath, N. P.; Srinivas, R. Synthesis 2003, 1895–1902. doi:10.1055/s-2003-41000
Return to citation in text: [1] -
Diels, O.; Alder, K. Liebigs Ann. Chem. 1928, 460, 98–122. doi:10.1002/jlac.19284600106
Return to citation in text: [1]
| 1. | Bindra, J. S. In The Alkaloids; Manske, R. H. F., Ed.; Academic Press: New York, 1973; Vol. 14, pp 84–121. |
| 14. | Liang, B.; Kalidindi, S.; Porco, J. A., Jr.; Stephenson, C. R. J. Org. Lett. 2010, 12, 572–575. doi:10.1021/ol902764k |
| 12. | Beeler, A. B.; Su, S.; Singleton, C. A.; Porco, J. A., Jr. J. Am. Chem. Soc. 2007, 129, 1413–1419. doi:10.1021/ja0674744 |
| 13. | Balthaser, B. R.; Maloney, M. C.; Beeler, A. B.; Porco, J. A., Jr.; Snyder, J. K. Nat. Chem. 2011, 3, 969–973. doi:10.1038/nchem.1178 |
| 9. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 18, 3003–3025. doi:10.1055/s-0029-1216975 |
| 10. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 11. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050 |
| 2. | Onishi, T.; Sebahar, P. R.; Williams, R. M. Tetrahedron 2004, 60, 9503–9515. doi:10.1016/j.tet.2004.07.047 |
| 3. | Onishi, T.; Sebahar, P. R.; Williams, R. M. Org. Lett. 2003, 5, 3135–3137. doi:10.1021/ol0351910 |
| 4. | Sebahar, P. R.; Williams, R. M. J. Am. Chem. Soc. 2000, 122, 5666–5667. doi:10.1021/ja001133n |
| 5. | Alper, P. B.; Meyers, C.; Lerchner, A.; Siegel, D. R.; Carreira, E. M. Angew. Chem., Int. Ed. 1999, 38, 3186–3189. doi:10.1002/(SICI)1521-3773(19991102)38:21<3186::AID-ANIE3186>3.0.CO;2-E |
| 6. | Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N. J. Am. Chem. Soc. 1999, 121, 2147–2155. doi:10.1021/ja983788i |
| 7. | Matsuura, T.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6500–6503. doi:10.1021/ja980788+ |
| 8. | Ashimori, A.; Bachand, B.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6477–6487. doi:10.1021/ja980786p |
| 22. | Diels, O.; Alder, K. Liebigs Ann. Chem. 1928, 460, 98–122. doi:10.1002/jlac.19284600106 |
| 18. | Roche, S. P.; Porco, J. A., Jr. Angew. Chem., Int. Ed. 2011, 50, 4068–4093. doi:10.1002/anie.201006017 |
| 19. | Nair, V.; Devipriya, S.; Suresh, E. Tetrahedron 2008, 64, 3567–3577. doi:10.1016/j.tet.2008.01.106 |
| 20. | Yavari, I.; Mokhtarporyani-Sanandaj, A.; Moradi, L. Tetrahedron Lett. 2007, 48, 6709–6712. doi:10.1016/j.tetlet.2007.07.135 |
| 21. | Nair, V.; Sreekanth, A. R.; Abhilash, N.; Biju, A. T.; Devi, B. R.; Menon, R. S.; Rath, N. P.; Srinivas, R. Synthesis 2003, 1895–1902. doi:10.1055/s-2003-41000 |
| 15. | Yin, X.-G.; Liu, X.-Y.; Hu, Z.-P.; Yan, M. Org. Biomol. Chem. 2012, 10, 1506–1509. doi:10.1039/C2OB06995D |
| 16. | Deng, H.-P.; Wei, Y.; Shi, M. Org. Lett. 2011, 13, 3348–3351. doi:10.1021/ol201094f |
| 17. | Hari Babu, T.; Joseph, A. A.; Muralidharan, D.; Perumal, P. T. Tetrahedron Lett. 2010, 51, 994–996. doi:10.1016/j.tetlet.2009.12.082 |
© 2012 Dai et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)