Abstract
The synthesis of a library of tetrahydro-β-carboline-containing compounds in milligram quantities is described. Among the unique heterocyclic frameworks are twelve tetrahydroindolizinoindoles, six tetrahydrocyclobutanindoloquinolizinones and three tetrahydrocyclopentenoneindolizinoindolones. These compounds were selected from a virtual combinatorial library of 11,478 compounds. Physical chemical properties were calculated and most of them are in accordance with Lipinski’s rules. Virtual docking and ligand-based target evaluations were performed for the β-carboline library compounds and selected synthetic intermediates to assess the therapeutic potential of these small organic molecules. These compounds have been deposited into the NIH Molecular Repository (MLSMR) and may target proteins such as histone deacetylase 4, endothelial nitric oxide synthase, 5-hydroxytryptamine receptor 6 and mitogen-activated protein kinase 1. These in silico screening results aim to add value to the β-carboline library of compounds for those interested in probes of these targets.

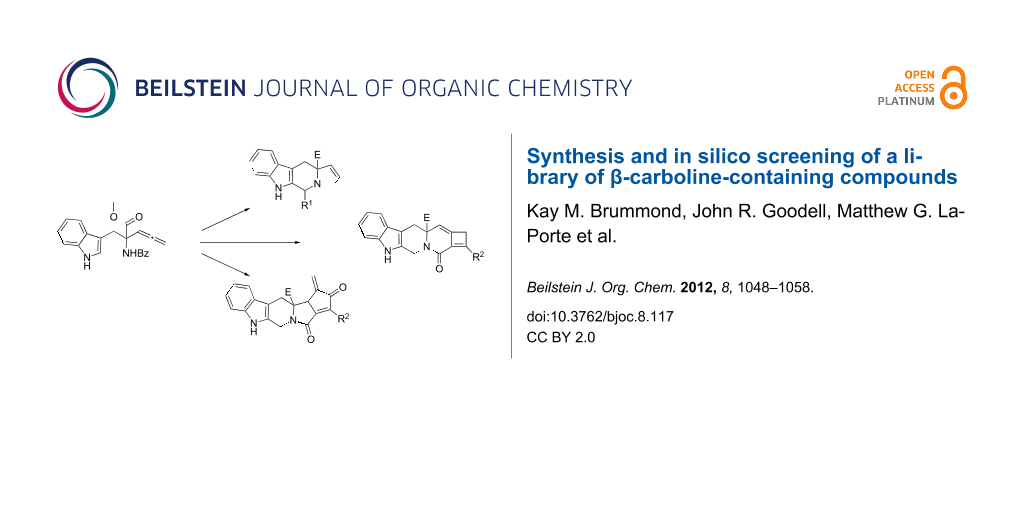
Graphical Abstract
Introduction
Identification of a comprehensive set of small organic molecules capable of selectively modifying the function of biological targets tremendously impacts modern medical research and drug discovery efforts [1]. Currently, this set of small molecules is largely occupied by in-house libraries and commercially available compounds. The NIH Roadmap initiative was established to address a recognized limitation of current compound diversity resulting in the Molecular Libraries Probe Centers Network (MLPCN) which has, since its inception, garnered a library of over 370,000 chemically diverse small molecules in a central molecule repository [2]. This supply of compounds has been made possible by researchers across the disciplines, but largely by synthetic chemists who are preparing compounds with an eye towards biologically relevant targets. Another goal of the NIH Roadmap is the development of enabling methods for the synthesis of these structurally diverse compound libraries; amongst these methods, skeletal diversification strategies have emerged as particularly efficient for maximizing structural diversity [3].
Previous work in the Brummond laboratory has demonstrated that an allene-containing β-carboline provided a good starting point for synthesizing six novel types of hetero-frameworks, all skeletally unique [4]. Moreover, scope and limitation studies contributed to an understanding of chemistries that would possess the robustness necessary for library preparation. Information gained from these experiments was then utilized in the construction of a virtual library of 11,748 compounds. A diversity analysis was performed using B (Burden) C (CAS) UT (Pearlman at the University of Texas) metrics and Tanimoto coefficients (Tc) and this virtual compound library was mapped onto the existing chemical space of the NIH Molecular Libraries Small Molecule Repository (MLSMR) [4]. When considering the physical properties most important to bimolecular interactions, atomic Gasteiger–Hückel charges, polarizabilities, and hydrogen-bond acceptors, these virtual compounds were found to occupy new chemical space when compared to the 327,000 compounds in the MLSMR. A small subset of these compounds was subsequently identified as ones representing a maximally diverse chemical space. The synthesis of a modified subset of this virtual compound library is described within, where modifications were mainly driven by studies of compound stability. Furthermore, a high throughput, in silico screening analysis of this library identified a number of potential biological targets for the compounds.
Results and Discussion
Scaffolds 1, 2 and 3 (Figure 1) were chosen for library preparation based upon favorable Tanimoto coefficient (Tc) scores when compared to the MLSMR, conformational constraints imposed by the β-carboline moiety, and the number of building blocks available for the diversifying elements R1 and R2.
![[1860-5397-8-117-1]](/bjoc/content/figures/1860-5397-8-117-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Tetrahydro-β-carboline containing scaffolds 1–3.
Figure 1: Tetrahydro-β-carboline containing scaffolds 1–3.
The syntheses of tetrahydro-β-carbolines 6{1–16} were accomplished in a manner entirely analogous to that reported previously (Table 1, entries 1–16) [4]. For example, the allenic methyl ester of tryptophan 4 was reacted with a number of aldehydes 5{1–15} under acidic conditions to produce the corresponding products in yields ranging from 54–89%. A range of aldehydes were accommodated in the Pictet–Spengler reaction, including formaldehyde (Table 1, entry 1), alkyl aldehydes (Table 1, entries 2 and 3), aryl aldehydes with electron-withdrawing and electron-donating groups (Table 1, entries 4–7, 14 and 15), heteroaromatic aldehydes (Table 1, entries 8–13) and glyoxalates (Table 1, entry 16). Moreover, useful quantities of β-carboline-containing products were obtained (43–100 mg). For entries 2–15, mixtures of two diastereomers were obtained. Since the mixtures could not be readily separated by column chromatography, diastereomeric ratios were determined by 1H NMR and were advanced without further purification. Reaction of allene 6 under the silver-nitrate-mediated cyclization conditions afforded the desired fused pyrrolines 1. However, in the initial phases of this cyclization process, a color change was noted during the purification process. Indeed, when NMR stability studies were performed on the syn- and anti-pyrrolines 1{5}, decomposition of both diastereomers was evident. Although it was generally difficult to isolate the individual diastereomers, they could be separated by column chromatography. It was found that anti-1{5} decomposed more rapidly than syn-1{5} during the 1H NMR stability studies, when compared to an internal standard. These results combined with previously reported skeletal reorganization processes of functionalized β-carbolines, led to concerns about the long-term storage of these compounds and their inclusion in the MLSMR [5].
Table 1: Library of N-tosyltetrahydro-β-carbolinepyrrolines 7{1–7}.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-117-i1.svg?max-width=637&scale=1.0)
|
||||||
| entry | R1 | yield of 6a (%) | anti:synb | yield of 7a (%) | purityc | |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-117-i2.svg?max-width=637&scale=1.0)
|
5{1} | 68 6{1} | NA | 28 7{1} | 98% |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-117-i3.svg?max-width=637&scale=1.0)
|
5{2} | 74 6{2} | 2.5:1 | 17 syn-7{2} | 98% |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-117-i4.svg?max-width=637&scale=1.0)
|
5{3} | 81 6{3} | 2.1:1 | 23 syn-7{3} | 98% |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-117-i5.svg?max-width=637&scale=1.0)
|
5{4} | 73 6{4} | 2.4:1 |
11 anti-7{4}
28 syn-7{4} |
98%
98% |
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-117-i6.svg?max-width=637&scale=1.0)
|
5{5} | 75 6{5} | 2.4:1 |
34 anti-7{5}
17 syn-7{5} |
98%
98% |
| 6 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-117-i7.svg?max-width=637&scale=1.0)
|
5{6} | 74 6{6} | 1.3:1 |
35 anti-7{6}
10 syn-7{6} |
98%
98% |
| 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-117-i8.svg?max-width=637&scale=1.0)
|
5{7} | 54 6{7} | 2.7:1 |
12 anti-7{7}
17 syn-7{7} |
98%
98% |
| 8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-117-i9.svg?max-width=637&scale=1.0)
|
5{8} | 89 6{8} | 2.3:1 | NDd | |
| 9 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-117-i10.svg?max-width=637&scale=1.0)
|
5{9} | 78 6{9} | 1.2:1 | NDe | |
| 10 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-117-i11.svg?max-width=637&scale=1.0)
|
5{10} | 80 6{10} | 1:1.1 | NDd | |
| 11 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-117-i12.svg?max-width=637&scale=1.0)
|
5{11} | 70 6{11} | 1.3:1 | NDe | |
| 12 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-117-i13.svg?max-width=637&scale=1.0)
|
5{12} | 80 6{12} | 3.5:1 | NDe | |
| 13 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-117-i14.svg?max-width=637&scale=1.0)
|
5{13} | 63 6{13} | 1:1.4 | NDe | |
| 14 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-117-i15.svg?max-width=637&scale=1.0)
|
5{14} | 58 6{14} | 2.3:1 | NDd | |
| 15 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-117-i16.svg?max-width=637&scale=1.0)
|
5{15} | 80 6{15} | 2.3:1 | NDd | |
| 16 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-117-i17.svg?max-width=637&scale=1.0)
|
5{16} | 88 6{16} | 1.4:1 | 8 syn-1{16} | 99% |
aIsolated yields; bdr determined by 1H NMR; cpurity established by LCMS/ELS; dcompound 6 detected; ecompound 6 not detected.
To increase the stability of this class of compounds, a toluenesulfonyl group was added to the indole nitrogen of 1{1–7} to give N-tosyl-tetrahydro-β-carbolinepyrroline derivatives 7{1–7}. These tosylated derivatives exhibited improved stability as evidenced by 1H NMR (Table 1, entries 1–7, and Supporting Information File 1, S76–S81). Incorporation of the tosyl group also eased the chromatographic separation of the syn- and anti-isomers for entries 2–7, thus compounds 7{2–7} were obtained as single diastereomers. Low to moderate yields for this two-step reaction sequence were attributed to a problematic tosylation due to the sterically hindered nature of the indole nitrogen atom. Moreover, unforeseen limitations were encountered for the heteroaromatic and naphthyl-containing β-carboline intermediates (Table 1, entries 8–15). While in some cases the intermediate pyrrolines 1 were observed (Table 1, entries 8, 10, 14, 15), the corresponding tosylated products were not obtained. The heteroaromatic examples (Table 1, entries 9, 11–13), did not undergo cyclization upon treatment with silver nitrate. For these cases, it was assumed that competing coordination of the heteroatom to the silver ion was an issue; however, attempts were not made to alter the reaction conditions for these substrates. Furthermore, conversion of the naphthalene-containing analogues 1{14} and 1{15} to their corresponding tosylates was not successful.
Next, compounds possessing the cyclobutene-fused β-carboline skeleton were assembled from the versatile allenyl intermediate 6. For this subset of compounds, acylation of the sterically hindered amine of 6{1} with the ynoic acids 8{1–8}, by using bromo-tris-pyrrolidino-phosponium hexafluorophosphate (PyBroP), provided the requisite allene-yne substrates 9{1,2}–9{1,8}. For the coupling reaction of the ynoic acids with an aryl group on the terminus of the alkyne 8{5–8}, the desired allene-ynes 9{1,5}–9{1,8} were afforded along with the [2 + 2] cycloadducts 2{1,5}–2{1,8} (Table 2, entries 5–8). Previous studies with related tryptophan-substituted allene-ynes, required much higher temperatures (225 °C versus rt) to give the [2 + 2] cycloadducts, albeit none of these examples possessed two radical stabilizing groups on the alkyne. Because the calculations performed by Tantillo [6] regarding the thermal [2 + 2] cycloaddition reaction suggest that the energy barrier for this reaction should not be effected by the presence of two radical stabilizing groups over one, it was postulated that this cycloaddition process was facilitated by exposure to incident light [7]. Thus, the mixtures of compounds 2 and 9 were reconstituted in CH2Cl2 and placed in front of two 6 W UV lamps for 16 h at rt to afford the desired cyclobutenes (Method B, Table 2, entries 5–8). During optimization of the conditions for the [2 + 2] cycloaddition reaction (Method A), it was found that reducing the reaction temperature from 225 °C to 160 °C afforded cyclobutene 2{1,3} in 73% yield (Table 2, entry 3). Similarly, allene-yne 9{1,5} was subjected to the lower reaction temperature (Method A) to produce cyclobutene 2{1,5} in 57% yield (Table 2, entry 5).
Table 2: Library of tetrahydro-β-carbolinecyclobutenes 2{1,3}–2{1,8}.
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-117-i18.svg?max-width=637&scale=1.0)
|
||||||
| entry | R2 | yield of 9a (%) | methodb | yield of 2a (%) | purityc | |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-117-i19.svg?max-width=637&scale=1.0)
|
8{1} | 0 9{1,1} | — | — | — |
| 2 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-117-i20.svg?max-width=637&scale=1.0)
|
8{2} | 64 9{1,2} | A | NDd | — |
| 3 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-117-i21.svg?max-width=637&scale=1.0)
|
8{3} | 75e 9{1,3} | A | 73 2{1,3} | 99% |
| 4 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-117-i22.svg?max-width=637&scale=1.0)
|
8{4} | 71 9{1,4} | A | 57 2{1,4} | 99% |
| 5 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-117-i23.svg?max-width=637&scale=1.0)
|
8{5} |
9{1,5}
2{1,5} |
A
B |
57f 2{1,5}
40 2{1,5} |
99% |
| 6 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-117-i24.svg?max-width=637&scale=1.0)
|
8{6} |
9{1,6}
2{1,6} |
B | 41 2{1,6} | 99% |
| 7 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-117-i25.svg?max-width=637&scale=1.0)
|
8{7} |
9{1,7}
2{1,7} |
B | 30 2{1,7} | 99% |
| 8 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-117-i26.svg?max-width=637&scale=1.0)
|
8{8} | 9{1,8} | B | 33 2{1,8} | 99% |
aIsolated yield; bmethod A: μW, 160 °C, DMF, 10 min; method B: Placed in front of two 6 W UV lamps (245 nm), CH2Cl2, rt, 16 h, no stirring; cpurity established by LCMS/ELS; dND = not detected; eμW, 225 °C, DMF, 7 min, (39%); fseparated 9{1,5} (57% yield) from 2{1,5} (18% yield) after the coupling reaction then submitted 9{1,5} to method A to give 2{1,5} in 68% yield. This compound was recombined with the previously isolated 2{1,5} to afford a combined 57% yield.
For the final library scaffold, a small subset of α-methylenecyclopentenone-containing tetrahydro-β-carbolines was synthesized. These compounds contain a general substructure that has recently been shown to inhibit DNA damage checkpoints [8]. Allene-ynes 9{1,3}–9{1,4} undergo Pauson–Khand cyclocarbonylation reactions when treated with molybdenum hexacarbonyl in DMSO/toluene solutions (Table 3). Allene-yne 9{1,2} afforded a mixture of four compounds comprising two diastereomers of 3{1,2}, the 4-alkylidene cyclopentenone, resulting from the cyclocarbonylation reaction with the distal double of the allene, and a fourth compound, which could not be identified (see spectral data in Supporting Information File 1). Aryl-substituted alkynones 9{1,5}–9{1,8} were not available for the molybdenum-mediated cyclocarbonylation process due to competing [2 + 2] cycloaddition reactions (Table 2).
Table 3: Synthesis of α-methylenecyclopentenone library 3{1,3}–3{1,4}.
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-117-i27.svg?max-width=637&scale=1.0)
|
||||
| entry | R2 | yield of 3a (%) | purityb | |
|---|---|---|---|---|
| 1 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-8-117-i28.svg?max-width=637&scale=1.0)
|
9{1,3} | 48 3{1,3} | 99% |
| 2 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-8-117-i29.svg?max-width=637&scale=1.0)
|
9{1,4} | 46 3{1,4} | 99% |
aIsolated yields; bpurity determined by LCMS/ELS.
The majority of these β-carboline-containing products exhibit acceptable calculated physical–chemical properties in accordance with Lipinski’s rule of five (Figure 2) [9,10]. These favorable properties and structural novelty make these valuable candidates for deposition in the MLSCN for biological activity evaluation.
![[1860-5397-8-117-2]](/bjoc/content/figures/1860-5397-8-117-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Library of tetrahydro-β-carboline containing compounds 1–7 and calculated properties (amolecular weight; bhydrogen-bond donor/acceptors; ccLogP; dpolar surface area (PSA)).
Figure 2: Library of tetrahydro-β-carboline containing compounds 1–7 and calculated properties (amolecular we...
Diversity-oriented synthesis (DOS) has been employed to generate thousands of the organic compounds that have been deposited in the NIH molecular repository for medicinal chemistry research. Deciphering the therapeutic potential of this many compounds is a continuing challenge. By combining chemogenomics databases, such as Protein Data Base (PDB) and ChEMBL, it is possible to map new compounds into existing chemical space and to predict protein targets for new compounds, for which there are two complementary strategies that can be implemented. One is a structure-based docking strategy, in which a query compound is fit into a series of protein binding pockets to identify favorable compound–protein interactions. A second approach is a ligand-based strategy in which the structural similarities between a query compound and a collection of bioactive compounds are identified. In the present study, both of these strategies were used to predict potential targets of the newly synthesized library of β-carboline-containing compounds.
High-throughput docking studies for protein-target prediction of newly synthesized compounds
Molecular docking studies were performed with the 34 newly synthesized compounds, represented by scaffolds 1, 2, 3, and allenyl precursors 4, 6{1–16}, 9{1–4} to identify potential protein targets [11]. Protein structures were downloaded from the PDB [12] and the analysis was limited to a selection of the 607 proteins defined as “druggable” targets, in order to reduce computational time [13]. (The complete listing of these proteins and their PDB IDs are provided in Supporting Information File 2). The Surflex-dock module of the Sybyl software was employed for protein preparation and docking of the β-carboline library [14,15]. Water molecules and ligands were removed from the protein structures and the active site of each protein was defined by the corresponding residues around the cocrystallized ligands. In-house algorithms were used to evaluate ligand-docking efficiency, and docking scores were used to assess and rank the protein targets.
A portion of the protein-scoring matrix is illustrated in Figure 3. Several interesting results emerged from this in silico analysis: (1) Twenty of the new compounds have docking scores greater than 7.0, a number that can be mapped to Kd values less than 100 nM, for several protein targets; (2) six compounds, 7{7} and 2{1,4–1,8}, are predicted to be ligands for a single protein, human HDAC4 (PDBID:2vqq); (3) compounds 2{1,5} and 2{1,7} are predicted to be high-affinity ligands for 3-hydroxy-3-methylglutaryl-coenzyme A, reductase (PDBID:2q6b) and HDAC4, respectively, even though they are structurally different from the corresponding cocrystallized ligands HR2 and TGF; and (4) compounds 2{1,6} and 6{10} are predicted to be ligands for a total of 15 protein targets. This high number of potential protein targets may be due to the electronegative trifluoromethyl group on 2{1,6} and the effect it would have on the α,β-unsaturated amide and the purported bioactivity of the pyrazole group of 6{10}.
![[1860-5397-8-117-3]](/bjoc/content/figures/1860-5397-8-117-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Results of high-throughput docking analysis. Top: A docking-score matrix arranged by compound IDs and PDB IDs; bottom: Structures of known ligands HR2 and TGF and the newly synthesized compounds 2{1,5} and 2{1,7}. Docking scores larger than 7.0 are red colored and can be mapped to Kd values less than 100 nM. The corresponding protein names of PDB IDs and the full docking-score matrix are listed in Supporting Information File 1.
Figure 3: Results of high-throughput docking analysis. Top: A docking-score matrix arranged by compound IDs a...
Ligand-based strategy for target prediction
Ligand-based target prediction algorithms have been developed based upon an established medicinal chemistry principle that structurally similar compounds, with comparable physical properties, should convey related biological properties [16,17]. In this study, structural similarities were calculated between the compounds of the β-carboline library and the bioactive compounds in the well-annotated database, ChEMBL version 13, the largest publicly available compound-target database, containing 1,143,682 distinct compounds, 8,845 targets and 6,933,068 bioactivity entries from 44,682 publications and PubChem bioassays [18,19]. The Openbabel FP2 fingerprint was used as a descriptor to assess similarities between molecules [20]. Tanimoto coefficients were calculated between the compounds of the β-carboline library and the ChEMBL database, and only β-carboline compounds with a Tc greater than 0.60 were considered for bioactivity analysis. A lower Tc threshold was used to identify a larger number of bioactivity targets. Table 4 lists the most promising bioactive targets for the newly synthesized β-carbolines together with the structurally similar lead compounds in ChEMBL along with their reported potency and literature citations. Several interesting results emerge from the comparison study performed, including a number of targets that the compounds should be screened against, such as C–C chemokine receptor type 3, gamma-aminobutyric acid receptor subunit gamma-2, breast adenocarcinoma cells, 5-hydroxytryptamine receptor 6, angiotensin-converting enzyme, and DNA polymerase iota. Moreover, nine of the twelve compounds are represented by allene precursors, ones that were not originally considered in the diversity analysis.
Table 4: Potential targets of β-carbolines based upon bioactivity data in ChEMBL.
| target | CHEMBL compound |
bioactivity
type and reference |
our compound | similarity score |
|---|---|---|---|---|
| C–C chemokine receptor type 3 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-8-117-i30.svg?max-width=637&scale=1.0)
CHEMBL33838 |
IC50 = 325 nM [21] |
![[Graphic 31]](/bjoc/content/inline/1860-5397-8-117-i31.svg?max-width=637&scale=1.0)
A |
0.79 |
| Leishmania donovani |
![[Graphic 32]](/bjoc/content/inline/1860-5397-8-117-i32.svg?max-width=637&scale=1.0)
CHEMBL55830 |
IC50 = 1.42 µM [22] |
![[Graphic 33]](/bjoc/content/inline/1860-5397-8-117-i33.svg?max-width=637&scale=1.0)
4 |
0.80 |
| Acanthocheilonema viteae |
![[Graphic 34]](/bjoc/content/inline/1860-5397-8-117-i34.svg?max-width=637&scale=1.0)
CHEMBL44573 |
Activity = 94% [23] |
![[Graphic 35]](/bjoc/content/inline/1860-5397-8-117-i35.svg?max-width=637&scale=1.0)
6{5} |
0.86 |
| Gamma-aminobutyric acid receptor subunit gamma-2 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-8-117-i36.svg?max-width=637&scale=1.0)
CHEMBL358326 |
IC50 = 250 nM [24] |
![[Graphic 37]](/bjoc/content/inline/1860-5397-8-117-i37.svg?max-width=637&scale=1.0)
6{1} |
0.84 |
| 5-Hydroxytryptamine receptor 6 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-8-117-i38.svg?max-width=637&scale=1.0)
CHEMBL370935 |
Ki = 271.3 nM [25] |
![[Graphic 39]](/bjoc/content/inline/1860-5397-8-117-i39.svg?max-width=637&scale=1.0)
7{1} |
0.69 |
| 3-Hydroxyacyl-CoA dehydrogenase type-2 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-8-117-i40.svg?max-width=637&scale=1.0)
CHEMBL1382101 |
Potency = 31.6 µM PubChem AID:893 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-8-117-i41.svg?max-width=637&scale=1.0)
7{7} |
0.64 |
| Benzodiazepine receptors |
![[Graphic 42]](/bjoc/content/inline/1860-5397-8-117-i42.svg?max-width=637&scale=1.0)
CHEMBL11901 |
Ki = 510 nM [26] |
![[Graphic 43]](/bjoc/content/inline/1860-5397-8-117-i43.svg?max-width=637&scale=1.0)
1{16} |
0.72 |
| Angiotensin-converting enzyme |
![[Graphic 44]](/bjoc/content/inline/1860-5397-8-117-i44.svg?max-width=637&scale=1.0)
CHEMBL148616 |
IC50 = 500 nM [27] |
![[Graphic 45]](/bjoc/content/inline/1860-5397-8-117-i45.svg?max-width=637&scale=1.0)
9{1,3} |
0.71 |
| Antithrombotic potency |
![[Graphic 46]](/bjoc/content/inline/1860-5397-8-117-i46.svg?max-width=637&scale=1.0)
CHEMBL1089460 |
IC50 = 8.56 nM [28] |
![[Graphic 47]](/bjoc/content/inline/1860-5397-8-117-i47.svg?max-width=637&scale=1.0)
9{1,4} |
0.69 |
| Mitogen-activated protein kinase 1 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-8-117-i48.svg?max-width=637&scale=1.0)
CHEMBL44295 |
Potency = 0.794 µM PubChem AID:995 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-8-117-i49.svg?max-width=637&scale=1.0)
6{4} |
0.82 |
| Breast adenocarcinoma cells |
![[Graphic 50]](/bjoc/content/inline/1860-5397-8-117-i50.svg?max-width=637&scale=1.0)
CHEMBL1650665 |
Inhibition = 78% [29] |
![[Graphic 51]](/bjoc/content/inline/1860-5397-8-117-i51.svg?max-width=637&scale=1.0)
6{6} |
0.79 |
| DNA polymerase iota |
![[Graphic 52]](/bjoc/content/inline/1860-5397-8-117-i52.svg?max-width=637&scale=1.0)
CHEMBL1360719 |
Potency = 1.78 µM PubChem AID:588590 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-8-117-i53.svg?max-width=637&scale=1.0)
6{7} |
0.85 |
Conclusion
A library of 34 β-carboline-containing compounds was synthesized utilizing a skeletal diversification strategy. High-throughput docking and ligand-based protocols were implemented to predict potential biological targets of the newly synthesized β-carbolines. The docking approach uses a structure-based technology to predict preferred interactions between compounds and protein targets, whereas the ligand-based method uses ligand similarity coefficients to identify potential biological targets. The complementary nature of these two protocols is evidenced by the fact that there was no overlap in the predicted biological targets. Furthermore, the in silico screening of these compounds is intended to add value to the library, by directing them to appropriate biological assays. Such strategies can also be used to explore the mechanisms of a biologically active compound in bioassays whose molecular target is as of yet unidentified.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectral data for compounds 1{5}, 1{16}, 2{1,3–8}, 3{1,2–4}, 4, 6{1–10}, 6{12–13}, 6{15}, 7{1–7}, 9{1,2–4}. | ||
| Format: PDF | Size: 17.8 MB | Download |
| Supporting Information File 2: The complete listing of the proteins and their PDB IDs (Targets Docking Score Matrix). | ||
| Format: XLS | Size: 582.5 KB | Download |
Acknowledgements
The authors are grateful to the NIH P50 GM067082 (118220) for generous financial support. This work is also partially supported by NIH R21HL109654 (Xie). We thank Drs. Lei Wang and Tom Jones at Tripos Company and Professor Peter Wipf at the University of Pittsburgh for professional discussions. Finally, the analytical support provided by Mr. Pete G. Chambers is very much appreciated.
References
-
Stockwell, B. R. Nature 2004, 432, 846–854. doi:10.1038/nature03196
Return to citation in text: [1] -
NIH Common Fund Office of Strategic Coordination. Molecular Libraries and Imaging. http://commonfund.nih.gov/molecularlibraries/overview.aspx (accessed June 14, 2012).
Return to citation in text: [1] -
Dandapani, S.; Marcaurelle, L. A. Curr. Opin. Chem. Biol. 2010, 14, 362–370. doi:10.1016/j.cbpa.2010.03.018
Return to citation in text: [1] -
Painter, T. O.; Wang, L.; Majumder, S.; Xie, X.-Q.; Brummond, K. M. ACS Comb. Sci. 2011, 13, 166–174. doi:10.1021/co100052s
Return to citation in text: [1] [2] [3] -
Diness, F.; Beyer, J.; Meldal, M. Chem.–Eur. J. 2006, 12, 8056–8066. doi:10.1002/chem.200600138
Return to citation in text: [1] -
Siebert, M. R.; Osbourn, J. M.; Brummond, K. M.; Tantillo, D. J. J. Am. Chem. Soc. 2010, 132, 11952–11966. doi:10.1021/ja102848z
Return to citation in text: [1] -
Murakami, M.; Matsuda, T. Cycloaddition of Allenes. In Modern Allene Chemistry; Krause, N.; Hashmi, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp 727–733.
Return to citation in text: [1] -
Huryn, D. M.; Brodsky, J. L.; Brummond, K. M.; Chambers, P. G.; Eyer, B.; Ireland, A. W.; Kawasumi, M.; LaPorte, M. G.; Lloyd, K.; Manteau, B.; Nghiem, P.; Quade, B.; Seguin, S. P.; Wipf, P. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6757–6762. doi:10.1073/pnas.1015251108
Return to citation in text: [1] -
Maestro 8.5.207-QikProp, 2.1; Schrödinger, LLC: New York.
For calculations of physicochemical properties.
Return to citation in text: [1] -
Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Adv. Drug Delivery Rev. 1997, 23, 3–25. doi:10.1016/S0169-409X(96)00423-1
Return to citation in text: [1] -
Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J. Nat. Rev. Drug Discovery 2004, 3, 935–949. doi:10.1038/nrd1549
Return to citation in text: [1] -
Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. Nucleic Acids Res. 2000, 28, 235–242. doi:10.1093/nar/28.1.235
Return to citation in text: [1] -
Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. BMC Bioinformatics 2008, 9, 104. doi:10.1186/1471-2105-9-104
Return to citation in text: [1] -
Jain, A. N. J. Med. Chem. 2003, 46, 499–511. doi:10.1021/jm020406h
Return to citation in text: [1] -
Jain, A. N. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. doi:10.1007/s10822-007-9114-2
Return to citation in text: [1] -
Martin, Y. C.; Kofron, J. L.; Traphagen, L. M. J. Med. Chem. 2002, 45, 4350–4358. doi:10.1021/jm020155c
Return to citation in text: [1] -
Xie, X.-Q.; Chen, J.-Z. J. Chem. Inf. Model. 2008, 48, 465–475. doi:10.1021/ci700193u
Return to citation in text: [1] -
Bender, A. Nat. Chem. Biol. 2010, 6, 309. doi:10.1038/nchembio.354
Return to citation in text: [1] -
Heikamp, K.; Bajorath, J. J. Chem. Inf. Model. 2011, 51, 1831–1839. doi:10.1021/ci200199u
Heikamp et al. carried out a large-scale similarity search investigation on 266 well-defined compound activity classes extracted from the ChEMBL database using molecular fingerprints. In 203 of 266 test cases (~76%), a compound recovery rate of at least 50% was observed.
Return to citation in text: [1] -
Guha, R.; Howard, M. T.; Hutchison, G. R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner, J.; Willighagen, E. L. J. Chem. Inf. Model. 2006, 46, 991–998. doi:10.1021/ci050400b
Return to citation in text: [1] -
Dhanak, D.; Christmann, L. T.; Darcy, M. G.; Jurewicz, A. J.; Keenan, R. M.; Lee, J.; Sarau, H. M.; Widdowson, K. L.; White, J. R. Bioorg. Med. Chem. Lett. 2001, 11, 1441–1444. doi:10.1016/S0960-894X(01)00248-7
Return to citation in text: [1] -
Singh, I. P.; Jain, S. K.; Kaur, A.; Singh, S.; Kumar, R.; Garg, P.; Sharma, S. S.; Arora, S. K. Eur. J. Med. Chem. 2010, 45, 3439–3445. doi:10.1016/j.ejmech.2010.04.033
Return to citation in text: [1] -
Srivastava, S. K.; Agarwal, A.; Chauhan, P. M. S.; Agarwal, S. K.; Bhaduri, A. P.; Singh, S. N.; Fatima, N.; Chatterjee, R. K. J. Med. Chem. 1999, 42, 1667–1672. doi:10.1021/jm9800705
Return to citation in text: [1] -
Lippke, K. P.; Schunack, W. G.; Wenning, W.; Mueller, W. E. J. Med. Chem. 1983, 26, 499–503. doi:10.1021/jm00358a008
Return to citation in text: [1] -
Cole, D. C.; Lennox, W. J.; Stock, J. R.; Ellingboe, J. W.; Mazandarani, H.; Smith, D. L.; Zhang, G.; Tawa, G. J.; Schechter, L. E. Bioorg. Med. Chem. Lett. 2005, 15, 4780–4785. doi:10.1016/j.bmcl.2005.07.028
Return to citation in text: [1] -
Cain, M.; Weber, R. W.; Guzman, F.; Cook, J. M.; Barker, S. A.; Rice, K. C.; Crawley, J. N.; Paul, S. M.; Skolnick, P. J. Med. Chem. 1982, 25, 1081–1091. doi:10.1021/jm00351a015
Return to citation in text: [1] -
Ksander, G. M.; de Jesus, R.; Yuan, A.; Ghai, R. D.; McMartin, C.; Bohacek, R. J. Med. Chem. 1997, 40, 506–514. doi:10.1021/jm960583g
Return to citation in text: [1] -
Liu, J.; Jiang, X.; Zhao, M.; Zhang, X.; Zheng, M.; Peng, L.; Peng, S. J. Med. Chem. 2010, 53, 3106–3116. doi:10.1021/jm901816j
Return to citation in text: [1] -
Mohamed, H. A.; Girgis, N. M. R.; Wilcken, R.; Bauer, M. R.; Tinsley, H. N.; Gary, B. D.; Piazza, G. A.; Boeckler, F. M.; Abadi, A. H. J. Med. Chem. 2011, 54, 495–509. doi:10.1021/jm100842v
Return to citation in text: [1]
| 4. | Painter, T. O.; Wang, L.; Majumder, S.; Xie, X.-Q.; Brummond, K. M. ACS Comb. Sci. 2011, 13, 166–174. doi:10.1021/co100052s |
| 14. | Jain, A. N. J. Med. Chem. 2003, 46, 499–511. doi:10.1021/jm020406h |
| 15. | Jain, A. N. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. doi:10.1007/s10822-007-9114-2 |
| 4. | Painter, T. O.; Wang, L.; Majumder, S.; Xie, X.-Q.; Brummond, K. M. ACS Comb. Sci. 2011, 13, 166–174. doi:10.1021/co100052s |
| 16. | Martin, Y. C.; Kofron, J. L.; Traphagen, L. M. J. Med. Chem. 2002, 45, 4350–4358. doi:10.1021/jm020155c |
| 17. | Xie, X.-Q.; Chen, J.-Z. J. Chem. Inf. Model. 2008, 48, 465–475. doi:10.1021/ci700193u |
| 3. | Dandapani, S.; Marcaurelle, L. A. Curr. Opin. Chem. Biol. 2010, 14, 362–370. doi:10.1016/j.cbpa.2010.03.018 |
| 12. | Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. Nucleic Acids Res. 2000, 28, 235–242. doi:10.1093/nar/28.1.235 |
| 2. | NIH Common Fund Office of Strategic Coordination. Molecular Libraries and Imaging. http://commonfund.nih.gov/molecularlibraries/overview.aspx (accessed June 14, 2012). |
| 13. | Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. BMC Bioinformatics 2008, 9, 104. doi:10.1186/1471-2105-9-104 |
| 7. | Murakami, M.; Matsuda, T. Cycloaddition of Allenes. In Modern Allene Chemistry; Krause, N.; Hashmi, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp 727–733. |
| 9. |
Maestro 8.5.207-QikProp, 2.1; Schrödinger, LLC: New York.
For calculations of physicochemical properties. |
| 10. | Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Adv. Drug Delivery Rev. 1997, 23, 3–25. doi:10.1016/S0169-409X(96)00423-1 |
| 6. | Siebert, M. R.; Osbourn, J. M.; Brummond, K. M.; Tantillo, D. J. J. Am. Chem. Soc. 2010, 132, 11952–11966. doi:10.1021/ja102848z |
| 11. | Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J. Nat. Rev. Drug Discovery 2004, 3, 935–949. doi:10.1038/nrd1549 |
| 5. | Diness, F.; Beyer, J.; Meldal, M. Chem.–Eur. J. 2006, 12, 8056–8066. doi:10.1002/chem.200600138 |
| 4. | Painter, T. O.; Wang, L.; Majumder, S.; Xie, X.-Q.; Brummond, K. M. ACS Comb. Sci. 2011, 13, 166–174. doi:10.1021/co100052s |
| 8. | Huryn, D. M.; Brodsky, J. L.; Brummond, K. M.; Chambers, P. G.; Eyer, B.; Ireland, A. W.; Kawasumi, M.; LaPorte, M. G.; Lloyd, K.; Manteau, B.; Nghiem, P.; Quade, B.; Seguin, S. P.; Wipf, P. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6757–6762. doi:10.1073/pnas.1015251108 |
| 21. | Dhanak, D.; Christmann, L. T.; Darcy, M. G.; Jurewicz, A. J.; Keenan, R. M.; Lee, J.; Sarau, H. M.; Widdowson, K. L.; White, J. R. Bioorg. Med. Chem. Lett. 2001, 11, 1441–1444. doi:10.1016/S0960-894X(01)00248-7 |
| 18. | Bender, A. Nat. Chem. Biol. 2010, 6, 309. doi:10.1038/nchembio.354 |
| 19. |
Heikamp, K.; Bajorath, J. J. Chem. Inf. Model. 2011, 51, 1831–1839. doi:10.1021/ci200199u
Heikamp et al. carried out a large-scale similarity search investigation on 266 well-defined compound activity classes extracted from the ChEMBL database using molecular fingerprints. In 203 of 266 test cases (~76%), a compound recovery rate of at least 50% was observed. |
| 20. | Guha, R.; Howard, M. T.; Hutchison, G. R.; Murray-Rust, P.; Rzepa, H.; Steinbeck, C.; Wegner, J.; Willighagen, E. L. J. Chem. Inf. Model. 2006, 46, 991–998. doi:10.1021/ci050400b |
| 28. | Liu, J.; Jiang, X.; Zhao, M.; Zhang, X.; Zheng, M.; Peng, L.; Peng, S. J. Med. Chem. 2010, 53, 3106–3116. doi:10.1021/jm901816j |
| 29. | Mohamed, H. A.; Girgis, N. M. R.; Wilcken, R.; Bauer, M. R.; Tinsley, H. N.; Gary, B. D.; Piazza, G. A.; Boeckler, F. M.; Abadi, A. H. J. Med. Chem. 2011, 54, 495–509. doi:10.1021/jm100842v |
| 26. | Cain, M.; Weber, R. W.; Guzman, F.; Cook, J. M.; Barker, S. A.; Rice, K. C.; Crawley, J. N.; Paul, S. M.; Skolnick, P. J. Med. Chem. 1982, 25, 1081–1091. doi:10.1021/jm00351a015 |
| 27. | Ksander, G. M.; de Jesus, R.; Yuan, A.; Ghai, R. D.; McMartin, C.; Bohacek, R. J. Med. Chem. 1997, 40, 506–514. doi:10.1021/jm960583g |
| 24. | Lippke, K. P.; Schunack, W. G.; Wenning, W.; Mueller, W. E. J. Med. Chem. 1983, 26, 499–503. doi:10.1021/jm00358a008 |
| 25. | Cole, D. C.; Lennox, W. J.; Stock, J. R.; Ellingboe, J. W.; Mazandarani, H.; Smith, D. L.; Zhang, G.; Tawa, G. J.; Schechter, L. E. Bioorg. Med. Chem. Lett. 2005, 15, 4780–4785. doi:10.1016/j.bmcl.2005.07.028 |
| 22. | Singh, I. P.; Jain, S. K.; Kaur, A.; Singh, S.; Kumar, R.; Garg, P.; Sharma, S. S.; Arora, S. K. Eur. J. Med. Chem. 2010, 45, 3439–3445. doi:10.1016/j.ejmech.2010.04.033 |
| 23. | Srivastava, S. K.; Agarwal, A.; Chauhan, P. M. S.; Agarwal, S. K.; Bhaduri, A. P.; Singh, S. N.; Fatima, N.; Chatterjee, R. K. J. Med. Chem. 1999, 42, 1667–1672. doi:10.1021/jm9800705 |
© 2012 Brummond et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)