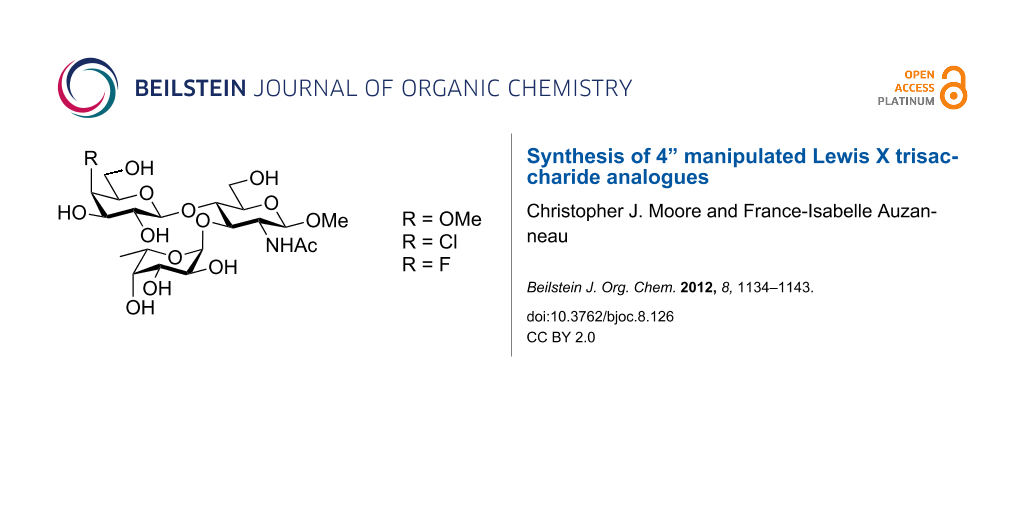
Abstract
Three analogues of the Lex trisaccharide antigen (β-D-Galp(1→4)[α-L-Fucp(1→3)]-D-GlcNAcp) in which the galactosyl residue is modified at O-4 as a methyloxy, deoxychloro or deoxyfluoro, were synthesized. We first report the preparation of the modified 4-OMe, 4-Cl and 4-F trichloroacetimidate galactosyl donors and then report their use in the glycosylation of an N-acetylglucosamine glycosyl acceptor. Thus, we observed that the reactivity of these donors towards the BF3·OEt2-promoted glycosylation at O-4 of the N-acetylglucosamine glycosyl acceptors followed the ranking 4-F > 4-OAc ≈ 4-OMe > 4-Cl. The resulting disaccharides were deprotected at O-3 of the glucosamine residue and fucosylated, giving access to the desired protected Lex analogues. One-step global deprotection (Na/NH3) of the protected 4”-methoxy analogue, and two-step deprotections (removal of a p-methoxybenzyl with DDQ, then Zemplén deacylation) of the 4”-deoxychloro and 4”-deoxyfluoro protected Lex analogues gave the desired compounds in good yields.

Graphical Abstract
Introduction
A glycolipid displaying the dimeric Lex hexasaccharide (dimLex) has been identified as a cancer associated carbohydrate antigen, particularly prevalent in colonic and liver adenocarcinomas. In addition, an association between the fucosylation of internal GlcNAc residues in polylactosamine chains, and metastasis and tumor progression in colorectal cancers has been suggested [1-6]. Unfortunately, dimLex displays the Lex trisaccharide (β-D-Galp(1→4)[α-L-Fucp(1→3)]-D-GlcNAcp), at the nonreducing end, and this antigenic determinant is also present at the surface of many normal cells and tissues, which include kidney tubules, gastrointestinal epithelial cells, and cells of the spleen and brain [7-11]. Indeed, anti-Lex monoclonal antibodies (e.g., FH3, SH1) have been shown to recognize this Lex antigenic determinant as it exists in the hexasaccharide [1-6]. Therefore, as our group embarks on the development of a therapeutic anticancer vaccine utilizing the Tumor Associated Carbohydrate Antigen (TACA) dimLex as a target, an important factor to consider is an expected autoimmune response to the native Lex antigen. Fortunately an internal epitope displayed by dimLex was discovered when monoclonal antibodies (mAbs) FH4 and SH2, raised against dimLex, were isolated. Indeed, these mAbs were shown to bind to the dimLex and trimLex antigens but only weakly recognise Lex trisaccharide antigen [1-6]. With this in mind, we focus our research on the discovery of analogues of dimLex that can be used as safe vaccine candidates. Ideally, these analogues should display the internal epitopes that are recognized by anti-dimLex SH2-like antibodies while being free of those that are recognized by anti-Lex SH1-like antibodies.
In order to abolish cross-reactivity with the Lex antigen, we have prepared [12-14] several analogues in an attempt to identify a suitable replacement for the nonreducing end Lex trisaccharide. In turn, we have compared the conformational behaviour of these analogues to that of the natural Lex-OMe 1 (Figure 1) through a mixture of stochastic searches and NMR analyses [15]. The results pointed toward the preferential adoption, by all of analogues, of the stacked conformation that has been assigned for the Lex trisaccharide [16-21]. The relative affinity of the anti-Lex monoclonal antibody SH1 [6] for the native Lex antigen 1 and our Lex analogues [12-14] was examined by competitive ELISA experiments using a Lex-BSA glycoconjugate as the immobilized ligand [15,22-24].
![[1860-5397-8-126-1]](/bjoc/content/figures/1860-5397-8-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of Lex and analogues 2–5.
Figure 1: Structure of Lex and analogues 2–5.
It was discovered that the Lex analogue 2, with a glucose unit in place of the galactose residue (Figure 1), did not bind to the SH1 mAb, even at high concentrations [15]. This discovery suggests that, to conserve cross-reactivity with the natural Lex antigen, the nonreducing end galactose is essential, and that modifying this residue, particularly at O-4, may lead to the complete loss of this cross-reactivity [15]. Currently, it is not known what the reason for this observed loss of binding is, since the binding affinities between proteins and carbohydrates are the result of a combination of factors [25-29]. One of the main interactions is the formation of hydrogen bonds, either direct or water-mediated, between the amino acid residues of the protein and the key binding hydroxy groups of the ligand, which are arranged in clusters presented by different monosaccharide units. Other factors include favourable interactions of the nonpolar amino acid residues with the hydrophobic patches exhibited by the ligand, as well as high-energy water molecules being favourably displaced from the combining site. Binding affinity is therefore a result of combined enthalpic, entropic and solvation effects, frequently leading to a balance between favourable enthalpic and unfavourable entropic contributions [25-29]. Thus, only additional competitive ELISA studies with Lex-OMe analogue inhibitors containing strategic manipulations at the 4” site will provide further insight into specific binding interactions [15]. The synthesis of a 4”-deoxy Lex trisaccharide analogue was reported recently by Dong et al. [30]. Here, we report the synthesis of 4”-methyloxy, 4”-deoxychloro and 4”-deoxyfluoro Lex-OMe analogue inhibitors 3–5.
Results and Discussion
There are numerous reports in the literature of the chemical preparation of Lex analogues [30-55], including one recent report of the synthesis of a Lex pentasaccharide 4-deoxy at the nonreducing end galactosyl residue [30]. These syntheses follow one of three strategies: (1) the galactosylation then fucosylation of a glucosamine acceptor [30-47]; (2) the fucosylation then galactosylation of a glucosamine acceptor [48-53]; or (3) the fucosylation at O-3 of a lactosamine derivative prepared from lactose [54,55]. Our synthetic approach to prepare analogues 3–5 followed the first strategy [30-47] and involved the use of N-acetylglucosamine glycosyl acceptors 6 [14] and 8, galactosyl donors 9–11, and known fucosyl donors 12 [56] and 13 [12] (Figure 2).
![[1860-5397-8-126-2]](/bjoc/content/figures/1860-5397-8-126-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Monosaccharide building blocks 6–13.
Figure 2: Monosaccharide building blocks 6–13.
Synthesis of monosaccharide building blocks 8–11
Glucosamine acceptor 8 was prepared in two steps from the known [14] benzylidene 7: the benzylidene acetal was first hydrolyzed, and then the resulting diol (79%) was selectively benzoylated at O-6 (BzCl-collidine) giving acceptor 8 in 62% yield.
The syntheses of trichloroacetimidate donors 9–11 are described on Scheme 1; they were all prepared from the known trichloroethyl galactoside 14 [57]. Galactoside 14 was first prepared in three steps from galactose: (1) peracetylation (Ac2O-pyridine); (2) BF3·OEt2 activation of the anomeric acetate and glycosidation with trichloroethanol; and (3) Zemplén deacetylation. This sequence of reactions gave the desired galactoside 14 in 78% yield and as a 9:1 α/β mixture, as assessed by 1H NMR. It is important to point out that the second step in this sequence of reactions used conditions very similar to those used by Risbood et al. to prepare peracetylated trichloroethyl galactopyranoside from peracetylated galactose. Indeed, in agreement with their work [57], the ratio of α-anomer isolated here suggests a late anomerization of the β-glycoside during our extended reaction time (20 h) at reflux. The 4-methyloxy trichloroacetimidate donor 9 was then prepared in four steps from the anomeric mixture of galactoside 14. Tetraol 14 was stirred in a mixture of pyridine and dichloromethane at −10 °C and treated with 3.1 equivalents of pivaloyl chloride. Under these conditions the α-tripivaloate 15, selectively acylated at O-2, O-3 and O-6, was obtained pure and free of β-anomer (64%). The free hydroxy group in alcohol 15 was then deprotonated with sodium hydride and allowed to react with methyl iodide, yielding the 4-OMe galactoside 16 in very good yield. In turn, trichloroethyl galactoside 16 was converted to the trichloroacetimidate donor 9 in two steps: the anomeric trichloroethyl group was removed (Zn/AcOH), and then the resulting hemiacetal was treated with trichloroacetonitrile in the presence of DBU giving the desired α-trichloroacetimidate in good yield.
![[1860-5397-8-126-i1]](/bjoc/content/inline/1860-5397-8-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of trichloroacetimidate donors 9–11.
Scheme 1: Synthesis of trichloroacetimidate donors 9–11.
A Lattrell-Dax nitrite mediated inversion [58-60] of the 4-OH in galactoside 15 provided the glucoside 17, which was used as the common precursor to analogues 18 and 19. Treatment with sulfuryl chloride [61] gave the 4-chloro galactoside 18 in good yield, whereas triflation at O-4 followed by SN2 displacement of the triflate by using tetrabutylammonium fluoride [62,63] gave the 4-fluoro galactoside analogue 19 in excellent yields. As described above for the preparation of donor 9 from glycoside 16, trichloroethyl galactosides 18 and 19 were, in turn, converted in two steps (Zn/AcOH then Cl3CCN/DBU) to the trichloroacetimidate donors 10 and 11, respectively, which were obtained in acceptable yields over the two steps.
Galactosylation at O-4 of N-acetylglucosamine acceptors
It has been well established that the hydroxy group at C-4 of N-acetylglucosamine is a poor nucleophile, with reduced reactivity toward glycosylation [64-66]. However, we have reported the successful O-4 glycosylation of an N-acetylglucosamine monosaccharide acceptor using peracetylated gluco- and galactopyranose α-trichloroacetimidate donors under activation with 2 equivalents of BF3·OEt2 at room temperature or 40 °C [14,67]. We applied similar conditions to prepare disaccharides 20–22 (Table 1). Glycosylation of methyl glycoside 6 with the 4-methoxy donor 9 gave the best results when the reaction was carried out at 40 °C and left to proceed for 2 hours. Under these conditions, the desired disaccharide 20 was isolated in acceptable yields (Table 1, entries 1 and 2). Our attempts to reduce the number of equivalents of donor 9 used in the reaction always resulted in a lower yield of the desired disaccharide. Glycosylation of acceptor 8 with the 4-chloro galactosyl donor 10 appeared to be slower (Table 1, entries 3–5) than that of acceptor 6 with donor 9. The best results were obtained when the reaction was left to proceed for 3 rather than 2 hours (Table 1, entry 4), and the desired disaccharide 21 was then obtained in acceptable yield. Increasing the temperature to 60 °C did not increase the yield, presumably due to the degradation of the glycosyl donor at this higher temperature (Table 1, entry 5). Of the three glycosylations considered here, the coupling of acceptor 8 with the 4-fluoro donor 11 gave the highest yields (Table 1, entries 6 and 7). Indeed the desired disaccharide 22 was obtained in very good yields when the reaction was allowed to proceed for 2 hours at 40 °C. Once again, increasing the temperature to 60 °C offered no advantage and in fact led to a lower yield of the desired product.
Table 1: Glycosylation at O-4 of glucosamine with donors 9–11.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-126-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Acc. | Don. | T (°C) | Time (h) | Product (%) |
|---|---|---|---|---|---|
| 1 | 6 | 9 | rtb | 2 | 20 (65) |
| 2 | 6 | 9 | 40b | 2 | 20 (70) |
| 3 | 8 | 10 | 40b | 2 | 21 (55) |
| 4 | 8 | 10 | 40b | 3 | 21 (63) |
| 5 | 8 | 10 | 60c | 2 | 21 (62) |
| 6 | 8 | 11 | 40b | 2 | 22 (77) |
| 7 | 8 | 11 | 60c | 2 | 22 (73) |
aReagents and conditions: BF3·Et2O (2 equiv), donor 9–11 (5 equiv); bsolvent: CH2Cl2; csolvent: 1,2-dichloroethane.
From these three reactions, it is clear that the substituent at O-4 of a galactosyl donor impacts the outcome of glycosylation at O-4 of N-acetylglucosamine acceptors. Indeed, we have previously observed that galactosylations of such acceptors [68,69] usually result in lower yields (~70%) than those for analogous glucosylations, which usually provided around 90% of the desired disaccharides [14,67]. The results described here indicate that 4-OAc galactosyl donors perform better than the 4-Cl donor 10, whereas the 4-OMe donor 9 performs as well as the 4-OAc analogues. In addition, of all of the galactosyl donors employed thus far in such reactions, the 4-fluorinated analogue seemed to perform the best. Thus the reactivity of galactosyl trichloroacetimidate donors towards the BF3·OEt2-promoted glycosylation at O-4 of N-acetylglucosamine glycosyl acceptors seems to follow the ranking 4-F > 4-OAc ≈ 4-OMe > 4-Cl.
Preparation of the protected Lex trisaccharide analogues
The synthesis of protected trisaccharide intermediates 26–28 is described in Scheme 2. First, acceptors 23–25 were prepared in good yields through the selective removal of the chloroacetate (thiourea) in disaccharides 20–22. Fucosylation of acceptor 23 with ethylthioglycoside 12 was first attempted under NIS and TMSOTf activation at low temperature (−30 °C). Under these conditions, the desired trisaccharide 26 was isolated in 78% yield but as an 8:2 mixture of the α and β-anomers as estimated by 1H NMR. Although the desired anomer 26α could be obtained pure upon purification by HPLC, it was isolated in a less than desirable yield of 48%. We thus attempted the coupling of acceptor 23 and donor 12 under activation with excess MeOTf (5 equiv). Indeed, we have reported that such conditions allow glycosylation at O-4 of N-acetylglucosamine acceptors through the in situ formation of the corresponding N-methylimidate, temporarily masking the N-acetyl group in the acceptor [70,71]. Thus, we expected a similar in situ formation of the methyl imidate in acceptor 23, which would further undergo fucosylation at O-3. However, since methylimidates are unstable when purified on silica gel, they were converted back to the acetamido before purification. Thus, once TLC had shown that all of the acceptor had been consumed, the reaction was worked up and the crude mixture was treated with Ac2O–AcOH prior to purification by chromatography [70,71]. Under these conditions, the desired trisaccharide 26α was obtained pure and free of the β-anomer in 77% yield.
![[1860-5397-8-126-i2]](/bjoc/content/inline/1860-5397-8-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of trisaccharides 26–28 and deprotection reactions giving 3–5.
Scheme 2: Synthesis of trisaccharides 26–28 and deprotection reactions giving 3–5.
Similar reaction conditions were applied for the glycosylation of acceptors 24 and 25 with ethylthioglycoside 13. Interestingly, these fucosylation reactions proved to be slower and required additional equivalents of donor 13 to proceed. However, after treatment of the reaction mixtures with AcOH–Ac2O, the desired trisaccharides 27 and 28 were isolated in good yields (Scheme 2) and exclusively as the α-fucosylated trisaccharides. As previously observed for other similar analogues [66,72,73], careful analysis of the 1H NMR spectra acquired for trisaccharides 26α, 27, 28 indicated that the glucosamine residue adopted a conformation distorted from the usual 4C1 chair conformation. The distorted conformations of the N-acetylglucosamine ring in analogues 26α, 27, 28 were characterized by vicinal coupling constants of 6.2–6.6 Hz measured between the ring hydrogens H-2 to H-5 of this residue, rather than the expected values of 8.0–8.3 Hz as observed, when measurable, for the same hydrogens in disaccharides 20–25. In addition, although signal overlap precluded its measurement in trisaccharides 26α and 28, the vicinal coupling constant measured between H-1 and H-2 in trisaccharide 27 (5.2 Hz) also supported a distorted conformation for this residue (compare to the same coupling constant in compounds 23–25, ~7.4 Hz).
Deprotection of trisaccharides 26–28
As described previously, the removal of various protecting groups, such as pivaloyl and benzyl groups here, can be accomplished efficiently in one step under Birch reduction conditions [15,68,69,74,75]. Thus, treatment of trisaccharide 26α with sodium in liquid ammonia at −78 °C was followed by neutralization of the reaction mixture with AcOH and purification by gel permeation chromatography on a Biogel P2 column (water) to give the desired deprotected 4”-methoxy trisaccharide analogue 3 pure in 83% yield. Since such conditions were not compatible with the 4-chloro and 4-fluoro substituents in trisaccharides 27 and 28, these intermediates were converted in two steps to the desired deprotected analogues 4 and 5, respectively. Thus, removal of the p-methoxybenzyl group with DDQ in CH2Cl2/H2O (15:1 v/v) was followed by Zemplén deacylation, giving the target Lex analogues 4 and 5 in 78% and 75%, respectively, over the two steps. The structures of the final deprotected trisaccharides 3–5 were confirmed by HR–ESI mass spectrometry and NMR.
Conclusion
We describe here the efficient synthesis of three Lex methyl glycoside derivatives (3–5) in which the galactosyl 4-hydroxy group is either methylated (3) or replaced by a chlorine (4) or fluorine (5). Our results seem to indicate that galactosylation at O-4 of an N-acetylglucosamine acceptor under activation with excess BF3·OEt2 can be significantly affected by the nature of the substituent present at C-4 of the galactosyl donor. Indeed, the best results were obtained with the 4-fluoro galactosyl donor, whereas the 4-chloro donor reacted less efficiently than the 4-O-methyl or 4-O-acetyl donors. Overall, this study also confirms our observation [68], that galactosylations at position 4 of N-acetylglucosamine acceptors are usually less successful than glucosylations [14,67]. The final Lex-OMe analogues will be used as competitive inhibitors in future ELISA experiments to provide a better understanding of the binding process between the anti-Lex monoclonal antibody SH1 and the Lex antigen.
Experimental
General Methods: 1H (400.13 MHz) and 13C NMR (100.6 MHz) spectra were recorded for compounds solubilized in CDCl3 (internal standard, for 1H: residual CHCl3 δ 7.24; for 13C: CDCl3 δ 77.0), DMSO-d6 (internal standard, for 1H: residual DMSO δ 2.54; for 13C: DMSO-d6 δ 40.45), CD3OD (internal standard, for 1H: residual MeOD δ 3.31; for 13C: CD3OD δ 49.15) or D2O [external standard 3-(trimethylsilyl)propionic acid-d4, sodium salt (TSP) for 1H δ 0.00; for 13C δ 0.00]. Chemical shifts and coupling constants were obtained from a first-order analysis of one-dimensional spectra. Assignments of proton and carbon resonances were based on COSY and 13C–1H heteronuclear correlated experiments. 1H NMR data are reported with standard abbreviations: singlet (s), doublet (d), triplet (t), doublet of doublet (dd), doublet of doublet of doublet (ddd), broad singlet (bs), broad triplet (bt), broad doublet of doublet (bdd) and multiplet (m). Mass spectra were obtained with electrospray ionization (ESI) on a high-resolution mass spectrometer. TLC were performed on precoated aluminum plates with Silica Gel 60 F254 and detected with UV light and/or by charring with a solution of 10% H2SO4 in EtOH. Compounds were purified by flash chromatography with Silica Gel 60 (230–400 mesh) unless otherwise stated. Solvents were distilled and dried according to standard procedures [76], and organic solutions were dried over Na2SO4 and concentrated under reduced pressure below 40 °C. HPLC purifications were run with HPLC grade solvents.
Methyl 2-acetamido-2-deoxy-3-O-(α-L-fucopyranosyl)-4-O-(4-methoxy-β-D-galactopyranosyl)-β-D-glucopyranoside (3). A solution of the protected trisaccharide 26α (50 mg, 0.043 mmol) dissolved in THF (5 mL) was added to a solution of liquid NH3 (ca. 20 mL) containing Na (72 mg, 3.13 mmol, 73 equiv) at −78 °C. After 1 h the reaction was quenched with MeOH (5 mL) and the ammonia was allowed to evaporate at rt. The remaining solution was neutralized with AcOH (220 μL, ca. 1.2 equiv to Na), and the mixture was concentrated. The resulting solid was dissolved in water and passed through a Biogel P2 column eluted with H2O. After freeze drying, the deprotected 4”-methoxy-Lex analogue 3 (20 mg, 0.0359 mmol, 83%) was obtained pure as a white solid. [α]D = −75 (c 0.3, H2O); 1H NMR (400 MHz, D2O, 295 K) δ 4.95 (d, J = 4.0 Hz, 1H, H-1’), 4.67 (m, 1H, H-5’), 4.32 (d, J = 8.2 Hz, 1H, H-1), 4.27 (d, J = 7.7 Hz, 1H, H-1”), 3.85 (dd, J = 2.0, 12.2 Hz, 1H, H-6a), 3.78–3.68 (m, 5H, H-2, H-3, H-4, H-3’, H-6b), 3.63–3.60 (m, 3H, H-4’, H-6ab”), 3.55–3.50 (m, 2H, H-2’, H-3”), 3.47–3.44 (m, 2H, H-5, H5”), 3.39 (d, J = 3.4 Hz, 1H, H-4”), 3.35 (s, 3H, OCH3), 3.32 (s, 3H, OCH3), 3.29 (m, 1H, H-2”), 1.88 (s, 3H, C(O)CH3), 1.03 (d, J = 6.6 Hz, 3H, H-6’); 13C NMR (100 MHz, D2O, 295 K) δ 174.4 (C=O), 101.7 (C-1), 101.6 (C-1”), 98.7 (C-1’), 78.6 (C-4”), 75.3, 75.1, 75.0 (C-5, C-5”, C-3), 73.3 (C-4), 72.9 (C-3”), 71.9 (C-4’), 71.5 (C-2”), 69.2 (C-3’), 67.7 (C-2’), 66.7 (C-5’), 61.1 (OCH3), 61.0 (C-6”), 59.7 (C-6), 57.1 (OCH3), 55.6 (C-2), 22.2 (C(O)CH3), 15.2 (C-6’); HRMS–ESI (m/z): [M + Na]+ calcd for C22H39NNaO15, 580.2217; found, 580.2223.
Methyl 2-acetamido-2-deoxy-4-O-(4-chloro-4-deoxy-β-D-galactopyranoside)-3-O-(α-L-fucopyranosyl)-β-D-glucopyranoside (4). A solution of the protected trisaccharide 27 (39 mg, 0.0347 mmol) and DDQ (12 mg, 1.5 equiv) in CH2Cl2 (350 μL) and H2O (20 μL, 6% v/v) was stirred at room temperature for 2 h. The mixture was filtered over Celite and the solids were washed with CH2Cl2 (5 mL). The combined filtrate and washings were washed with aq saturated NaHCO3 (10 mL), the aq layer was re-extracted with CH2Cl2 (3 × 5 mL), and the combined organic layers were dried and concentrated. Flash chromatography (EtOAc/hexanes, 1:1 → 7:3) of the residue gave a white solid (27 mg, 0.0269 mmol, 78%), which was dissolved in a methanolic solution of NaOMe (1 mL, 0.13 M) and stirred for 3 h at 50 °C. The reaction mixture was diluted with MeOH (5 mL) and de-ionized with DOWEX H+ resin. The resin was filtered off and washed with MeOH (5 mL), and the combined filtrated and washings were concentrated. The crude product was dissolved in Milli Q water and washed with CH2Cl2 (5 mL). After freeze drying, the deprotected 4”-deoxychloro Lex analogue 4 (15.1 mg, 0.0269 mmol, 78%) was obtained pure as an amorphous solid. [α]D = −123 (c = 0.7, H2O); 1H NMR (400 MHz, D2O, 295 K) δ 4.97 (d, 1H, J = 4.0 Hz, H-1’), 4.61 (m, 1H, H-5’), 4.38–4.30 (m, 3H, H-1, H-1”, H-4”), 3.88 (dd, J = 1.9, 12.3 Hz, 1H, H-6a), 3.82 (dd, J = 3.8, 9.7 Hz, 1H, H-3”), 3.77–3.70 (m, 7H, H-2, H-3, H-4, H-6b, H-3’, H-4’, H-5”), 3.66 (dd, J = 7.1, 11.7 Hz, 1H, H-6a”), 3.59–3.53 (m, 2H, H-2’, H-6b”), 3.45 (m, 1H, H-5), 3.38–3.33 (m, 4H, H-2”, OCH3), 1.88 (s, 3H, C(O)CH3), 1.05 (d, J = 6.6 Hz, 3H, H-6’); 13C NMR (100 MHz, D2O, 295 K) δ 174.4 (C=O), 102.2 (C-1”), 101.7 (C-1), 98.5 (C-1’), 75.2 (C-5), 74.4 (C-3), 73.7 (C-4), 73.6 (C-5”), 71.9 (C-4’), 71.5 (C-3”), 70.8 (C-2”), 69.3 (C-3’), 67.6 (C-2’), 66.6 (C-5’), 62.2 (C-4”), 61.6 (C-6”), 59.5 (C-6), 57.1 (OCH3), 55.7 (C-2), 22.2 (C(O)CH3), 15.2 (C-6’); HRMS–ESI (m/z): [M + Na]+ calcd for C21H36ClNNaO14, 584.1722; found, 584.1733.
Methyl 2-acetamido-2-deoxy-4-O-(4-deoxy-4-fluoro-β-D-galactopyranoside)-3-O-(α-L-fucopyranosyl)-β-D-glucopyranoside (5). Trisaccharide 28 (30 mg, 0.0271 mmol) was deprotected in two steps as described above for the preparation of trisaccharide 4. After freeze drying, the deprotected 4”-deoxyfluoro Lex analogue 5 (11.1 mg, 0.0203 mmol, 75%) was obtained pure as an amorphous solid. [α]D = −85 (c 0.5, H2O); 1H NMR (400 MHz, D2O, 295 K) δ 4.95 (d, J = 4.0 Hz, 1H, H-1’), 4.66 (m, 1H, H-5’), 4.65 (bdd, J = 2.7 Hz, JH,F = 50.4 Hz, 1H, H-4”), 4.39 (d, J = 7.8 Hz, 1H, H-1”), 4.33 (d, J = 8.0 Hz, 1H, H-1), 3.86 (dd, J = 2.0, 12.3 Hz, 1H, H-6a), 3.81–3.66 (m, 6H, H-2, H-3, H-4, H-6b, H-3’, H-3”), 3.64–3.58 (m, 4H, H-4’, H-5”, H-6ab”), 3.54 (dd, J = 4.0, 10.4 Hz, 1H, H-2’), 3.45 (m, 1H, H-5), 3.37–3.33 (m, 4H, H-2”, OCH3), 1.88 (s, 3H, C(O)CH3), 1.02 (d, J = 6.6 Hz, 3H, H-6’); 13C NMR (100 MHz, D2O, 295 K) δ 174.4 (C=O), 101.7 (C-1), 101.4 (C-1”), 98.7 (C-1’), 89.3 (d, JC,F = 177.7 Hz, C-4”), 75.2 (C-5), 74.9 (C-3), 73.4 (C-4), 73.2 (d, JC,F 17.6 Hz, C-5”), 71.9 (C-4’), 71.2 (C2”), 71.1 (d, JC,F = 18.5 Hz, C-3”), 69.2 (C-3’), 67.6 (C-2’), 66.5 (C-5’), 60.0 (C-6”), 59.6 (C-6), 57.1 (OCH3), 55.6 (C-2), 22.2 (C(O)CH3), 15.3 (C-6’); HRMS–ESI (m/z): [M + Na]+ calcd for C21H36FNO14, 568.2018; found, 568.2023.
Methyl 2-acetamido-6-O-benzyl-3-O-chloroacetyl-2-deoxy-4-O-(4-O-methyl-2,3,6-tri-O-pivaloyl-β-D-galactopyranosyl)-β-D-glucopyranoside (20). A solution of acceptor 6 (215 mg, 0.535 mmol) and trichloroacetimidate donor 9 (1.58 g, 5.0 equiv) in CH2Cl2 (30 mL) was stirred at 40 °C, and BF3·OEt2 (134 μL, 2.0 equiv) was added. The reaction was allowed to proceed for 2 h at 40 °C and then quenched with Et3N (179 μL, 2.4 equiv), and the mixture was diluted with CH2Cl2 (70 mL). The mixture was washed with aq saturated NaHCO3 (100 mL), the aq layer was re-extracted with CH2Cl2 (20 mL × 3), and the combined organic layers were dried and concentrated. Flash chromatography (EtOAc/hexanes, 2:8 → 6:4) of the residue gave disaccharide 20 (312 mg, 0.375 mmol, 70%) pure as a colourless glass. [α]D = −11 (c 0.6, CH2Cl2); 1H NMR (400 MHz, CDCl3, 296 K) δ 7.29 (m, 5H, Harom), 5.95 (d, J = 9.2 Hz, 1H, NH), 5.08–4.99 (m, 2H, H-3, H-2’), 4.71–4.68 (m, 2H, H-3’, CHHPh), 4.40 (d, J = 12.1 Hz, 1H, CHHPh), 4.38 (d, J = 7.4 Hz, 1H, H-1), 4.24–4.19 (m, 2H, H-1’, H-6a’), 4.13–4.07 (m, 2H, H-6b’, CHHCl), 4.01 (d, J = 15.1 Hz, 1H, CHHCl), 3.96–3.92 (m, 2H, H-4, H-2), 3.71 (m, 2H, H-6ab), 3.50–3.40 (m, 3H, H-5, H-4’, H-5’), 3.43, 3.41 (2s, 6H, 2 × OCH3), 1.94 (s, 3H, C(O)CH3), 1.30, 1.15, 1.10 (3s, 27H, 3 × C(CH3)3); 13C NMR (100 MHz, CDCl3, 296 K) δ 177.9, 177.7, 176.2, 170.3, 167.3 (C=O), 137.7, 128.6, 128.1, 128.0 (Ar), 101.7 (C-1), 99.2 (C-1’), 76.3 (C-4’), 74.2 (C-5), 73.9 (C-3), 73.5 (CH2Ph), 73.4 (C-3’), 72.1 (C-4), 72.0 (C-5’), 69.5 (C-2’), 67.7 (C-6), 61.7 (C-6’), 61.5 (OCH3), 56.6 (OCH3), 52.6 (C-2), 40.8 (CH2Cl), 38.8, 38.7, 38.6 (C(CH3)3), 27.2, 27.1 (C(CH3)3), 23.3 (C(O)CH3); HRMS–ESI (m/z): [M + H]+ calcd for C40H61ClNO15, 830.3730; found, 830.3735.
Methyl 2-acetamido-6-O-benzoyl-3-O-chloroacetyl-4-O-(4-chloro-4-deoxy-2,3,6-tri-O-pivaloyl-β-D-galactopyranosyl)-2-deoxy-β-D-glucopyranoside (21). Glycosylation of acceptor 8 (97 mg, 0.233 mmol) with trichloroacetimidate 10 (694 mg, 5.0 equiv) was performed under BF3·OEt2 (59 μL, 2.0 equiv) activation as described above for the synthesis of disaccharide 20. Work-up, as described above, and flash chromatography (EtOAc/hexanes, 2:8 → 6:4) of the residue gave disaccharide 21 (125 mg, 0.147 mmol, 63%) pure as a colourless glass. [α]D = +9 (c 0.9, CH2Cl2); 1H NMR (400 MHz, CDCl3, 295 K) δ 8.00–7.41 (m, 5H, Ar), 5.87 (d, J = 9.3 Hz, 1H, NH), 5.27–5.16 (m, 2H, H-2’, H-3), 4.86 (dd, J = 3.9, 10.1 Hz, 1H, H-3’), 4.61 (dd, J = 2.9, 12.0 Hz, 1H, H-6a), 4.51–4.47 (m, 3H, H-1, H-6b, H-1’), 4.38 (d, J = 3.5 Hz, 1H, H-4’), 4.35–4.30 (dd, J = 7.2, 11.5 Hz, 1H, H-6a’), 4.16–4.02 (m, 4H, H-2, H-6b’, CH2CCl3), 3.91 (t, J = 8.3 Hz, 1H, H-4), 3.83 (bt, J = 6.2 Hz, 1H, H-5’), 3.72 (m, 1H, H-5), 3.45 (s, 3H, OCH3), 1.97 (s, 3H, C(O)CH3), 1.16, 1.14, 1.13 (3s, 27H, 3 × C(CH3)3); 13C NMR (100 MHz, CDCl3, 295 K) δ 177.8, 177.6, 176.3, 170.3, 167.3, 166.0 (C=O), 133.5, 129.6, 129.4, 128.7, 128.4 (Ar), 101.7 (C-1), 100.4 (C-1’), 73.5 (C-3), 73.4 (C-4), 72.5 (C-5), 71.6 (C-3’), 71.2 (C-5’), 68.3 (C-2’), 62.6 (C-6), 62.6 (C-6’), 57.2 (C-4’), 56.8 (OCH3), 52.6 (C-2), 40.7 (CH2Cl), 38.9, 38.8, 38.7 (C(CH3)3), 27.6, 27.1, 27.0, 26.9, 26.7 (C(CH3)3), 23.3 (C(O)CH3); HRMS–ESI (m/z): [M + H]+ calcd for C39H56Cl2NO15, 848.3027; found, 848.3009.
Methyl 2-acetamido-6-O-benzoyl-3-O-chloroacetyl-2-deoxy-4-O-(4-deoxy-4-fluoro-2,3,6-tri-O-pivaloyl-β-D-galactopyranosyl)-β-D-glucopyranoside (22). Glycosylation of acceptor 8 (91.5 mg, 0.220 mmol) with trichloroacetimidate 11 (637 mg, 5.0 equiv) was performed under BF3·OEt2 (134 μL, 2.0 equiv) activation as described above for the synthesis of disaccharide 20. Work-up, as described above, and flash chromatography (EtOAc/hexanes, 2:8 → 6:4) of the residue gave disaccharide 22 (143 mg, 0.172 mmol, 77%) pure as a colourless glass. [α]D = +9 (c 2.2, CH2Cl2); 1H NMR (400 MHz, CDCl3, 295 K) δ 7.98–7.46 (m, 5H, Harom), 6.00 (d, J = 9.3 Hz, 1H, NH), 5.21–5.16 (m, 2H, H-3, H-2’), 4.82 (ddd, J = 2.6, 10.3 Hz, JH,F = 26.9 Hz, 1H, H-3’), 4.70 (dd, J = 2.6 Hz, JH,F = 42.9 Hz, 1H, H-4’), 4.62 (m, 1H, H-6a), 4.51–4.46 (m, 3H, H-1, H-6b, H-1’), 4.29 (dd, J = 7.6, 11.4 Hz, 1H, H-6a’), 4.16 (dd, J = 5.6, 11.5 Hz, 1H, H-6b’), 4.09–4.00 (m, 3H, H-2, CH2Cl), 3.92 (t, J = 8.2 Hz, 1H, H-4), 3.72 (m, 1H, H-5), 3.66 (dt, J = 6.4 Hz, JH,F = 25.8 Hz, 1H, H-5’), 3.44 (s, 3H, OCH3), 1.97 (s, 3H, OCH3), 1.16, 1.13 (2s, 27H, 3 × C(CH3)3); 13C NMR (100 MHz, CDCl3, 295 K) δ 177.8, 177.6, 176.5, 170.46, 167.3, 165.9 (C=O), 133.5, 129.5, 129.4, 128.6 (Ar), 101.6 (C-1), 99.9 (C-1’), 85.3 (d, JC,F = 186.4 Hz, C-4’), 73.5 (C-3), 73.4 (C-4), 72.4 (C-5), 71.2 (d, JC,F = 18.0 Hz, C-3’), 71.1 (d, JC,F = 18.0 Hz, C-5’), 68.6 (C-2’), 62.6 (C-6), 61.2 (C-6’), 56.8 (OCH3), 52.5 (C-2), 40.6 (CH2Cl), 38.8, 38.8, 38.7 (C(CH3)), 27.1, 26.9 (C(CH3)), 23.2 (C(O)CH3); HRMS–ESI (m/z): [M + H]+ calcd for C39H56ClFNO15, 832.3323; found, 832.3344.
Methyl 2-acetamido-6-O-benzyl-3-O-(2,3,4-tri-O-benzyl-α-L-fucopyranosyl)-2-deoxy-4-O-(4-O-methyl-2,3,6-tri-O-pivaloyl-β-D-galactopyranosyl)-β-D-glucopyranoside (26). A mixture of disaccharide acceptor 23 (30 mg, 0.0398 mmol), known [56] thioethyl fucopyranoside 12 (76 mg, 0.159 mmol, 4.0 equiv), and activated powdered 4 Å molecular sieves (0.25 g) in Et2O (3.0 mL, 0.13 M), was stirred for 1 h at rt under N2. Then, MeOTf (23 μL, 5.0 equiv) was added, the reaction mixture was stirred for 30 min, and the reaction quenched with Et3N (33 μL, 6.0 equiv). Solids were filtered off on Celite and washed with CH2Cl2 (20 mL), and the combined filtrate and washings were washed with aq saturated NaHCO3 (15 mL). The aq layer was re-extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were dried and concentrated. The residue was dissolved in 25% AcOH in Ac2O (5 mL), and the solution was stirred at rt for 12 h and co-concentrated with toluene (3 × 10 mL). Flash chromatography (EtOAc/hexanes, 2:8 → 1:1) of the residue gave trisaccharide 26 (35.7 mg, 0.0305 mmol, 77%) pure as a colourless glass. [α]D = −56 (c 0.7, CH2Cl2); 1H NMR (400 MHz, CDCl3, 296 K) δ 7.32–7.17 (m, 20H, Harom), 5.96 (d, J = 7.6 Hz, 1H, NH), 5.14 (dd, J = 8.1, 10.4 Hz, 1H, H-2”), 5.04 (d, J = 3.6 Hz, 1H, H-1’), 4.89 (d, J = 11.7 Hz, 1H, CHHPh), 4.78–4.66 (m, 6H, H-1, H-3”, 2 × CH2Ph), 4.60 (d, J = 11.7 Hz, 1H, CHHPh), 4.56 (d, J = 12.1 Hz, 1H, CHHPh), 4.31 (m, 2H, H-1”, CHHPh), 4.21 (m, 1H, H-5’), 4.10–4.00 (m, 4H, H-4, H-2’, H-6ab”), 3.89 (t, J = 6.3 Hz, 1H, H-3), 3.86 (dd, J = 2.6, 10.1 Hz, 1H, H-3’), 3.79 (dd, J = 4.9, 10.1 Hz, 1H, H-6a), 3.69 (dd, J = 3.7, 10.1 Hz, 1H, H-6b), 3.59 (bd, J = 1.4 Hz, 1H, H-4’), 3.46–3.40 (m, 4H, H-2, H-5, H-4”, H-5”), 3.29, 3.26 (2s, 6H, 2 × OCH3), 1.73 (s, 3H, C(O)CH3), 1.14–1.08 (m, 30H, H-6’, 3 × C(O)C(CH3)3); 13C NMR (100 MHz, CDCl3, 296 K) δ 177.7, 177.6, 176.8, 170.3 (C=O), 139.1, 139.0, 138.7, 138.0, 128.5–127.0 (Ar), 100.4 (C-1), 98.8 (C-1”), 96.2 (C-1’), 79.7 (C-3’), 77.0 (C-4’), 76.5 (C-4), 76.2 (C-4”), 74.7 (CH2Ph), 73.4 (C-5”), 73.3 (C-3”), 73.0, 72.6, 72.2 (3 × CH2Ph), 72.0 (C-3), 71.8 (C-2’), 71.6 (C-5), 69.1 (C-2”), 68.8 (C-6), 66.6 (C-5’), 61.5 (C-6”), 61.3 (OCH3), 56.5 (OCH3), 53.5 (C-2) 38.8, 38.7 (C(CH3)3), 27.2, 27.1 (C(CH3)3), 23.1 (C(O)CH3), 16.6 (C-6’); HRMS–ESI (m/z): [M + H]+ calcd for C65H88NO18, 1170.6001; found, 1170.6033.
Methyl 2-acetamido-3-O-(3,4-acetyl-2-O-paramethoxybenzyl-α-L-fucopyranosyl)-6-O-benzoyl-4-O-(4-chloro-4-deoxy-2,3,6-pivaloyl-β-D-galactopyranosyl)-2-deoxy-β-D-glucopyranoside (27). A mixture of disaccharide acceptor 24 (48 mg, 0.0622 mmol), known [12] thiophenyl fucopyranoside 13 (86 mg, 0.187 mmol, 3.0 equiv) and activated powdered 4 Å molecular sieves (0.3 g) in Et2O (2.0 mL) was stirred 1 h at rt under N2. MeOTf (35 μL, 5.0 equiv) was added and the reaction was allowed to proceed for 3 h at rt. More donor 13 (43 mg, 1.5 equiv) was added and the reaction was allowed to proceed for an additional 1 h at rt before being quenched with Et3N (52 μL, 6.0 equiv). Work up of the reaction and treatment of the crude product in 25% AcOH in Ac2O (6 mL), as well as the subsequent work-up, were carried out as described above for the synthesis of trisaccharide 26. Flash chromatography (EtOAc/hexanes, 2:8 → 6:4) of the residue gave trisaccharide 27 (42.5 mg, 0.0379 mmol, 61%) pure as a colourless glass. [α]D = –21 (c 0.8, CH2Cl2); 1H NMR (400 MHz, CDCl3, 295 K) δ 8.00–6.84 (m, 9H, Harom), 6.01 (d, J = 7.6 Hz, 1H, NH), 5.32–5.24 (m, 3H, H-3’, H-4’, H-2”), 5.18 (d, J = 3.6 Hz, 1H, H-1’), 4.90 (dd, J = 3.8, 10.0 Hz, 1H, H-3”), 4.80 (d, J = 5.2 Hz, 1H, H-1), 4.72 (dd, J = 3.6, 11.9 Hz, 1H,H-6a), 4.65–4.52 (m, 5H, H-6b, H-5’, H-1”, CH2Ph), 4.42 (bd, J = 3.4 Hz, 1H, H-4”), 4.39–4.34 (m, 2H, H-6ab”), 4.20 (t, J = 6.6 Hz, 1H, H-3), 3.96 (t, J = 6.4 Hz, 1H, H-4), 3.89 (dd, J = 3.7, 10.4 Hz, 1H, H-2’), 3.81–3.76 (m, 2H, H-5, H-5”), 3.76 (s, 3H, OCH3), 3.58 (m, 1H, H-2), 3.34 (s, 3H, OCH3), 2.11, 1.95, 1.86 (3s, 9H, 3 × C(O)CH3), 1.15–1.14 (m, 30H, H-6’, 3 × C(CH3)3); 13C NMR (100 MHz, CDCl3, 295 K) δ 177.6, 177.5, 170.5, 170.4, 169.7, 166.0, 159.2 (C=O), 133.4, 130.4, 129.7, 129.5, 128.9, 128.7 (Ar), 100.4 (C-1), 100.0 (C-1”), 96.1 (C-1’), 73.3 (C-4, C-2’), 72.6 (CH2Ph), 72.2 (C-5”), 71.7 (C-4’, C-3, C-5), 71.5 (C-3”), 70.4 (C-3’), 68.0 (C-2”), 65.0 (C-5’), 63.7 (C-6), 62.2 (C-6”), 57.6 (C-4”), 56.6, 55.3 (OCH3), 53.5 (C-2) 38.9, 38.8, 38.7 (C(CH3)3), 27.1, 27.0, 27.0 (C(CH3)3), 23.2, 20.9, 20.7 (C(O)CH3), 15.9 (C-6’); HRMS–ESI (m/z): [M + H]+ calcd for C55H77ClNO21, 1122.4677; found, 1122.4626.
Methyl 2-acetamido-3-O-(3,4-di-O-acetyl-2-O-p-methoxybenzyl-α-L-fucopyranosyl)-6-O-benzoyl-2-deoxy-4-O-(4-deoxy-4-fluoro-2,3,6-pivaloyl-β-D-galactopyranosyl)-β-D-glucopyranoside (28). A mixture of disaccharide acceptor 25 (24 mg, 0.0318 mmol), known [12] thiophenyl fucopyranoside 13 (44 mg, 0.0953 mmol, 3.0 equiv) and activated powdered 4 Å molecular sieves (0.15 g) in Et2O (1.5 mL) was stirred for 1 h at rt under N2. MeOTf (18 μL, 5.0 equiv) was added and the reaction was allowed to proceed for 30 min at rt. More donor 13 (44 mg, 3.0 equiv) was added and the reaction was allowed to proceed for an additional 2 h at rt before being quenched with Et3N (27 μL, 6.0 equiv). Work up of the reaction and treatment of the crude product in 25% AcOH in Ac2O (4 mL), as well as the subsequent work-up, were carried out as described above for the synthesis of trisaccharide 26. Flash chromatography (EtOAc/hexanes, 2:8 → 6:4) of the residue gave trisaccharide 28 (22.8 mg, 0.0206 mmol, 65%) pure as a colourless glass. [α]D = −37 (c 1.2, CH2Cl2); 1H NMR (400 MHz, CDCl3, 295 K) δ 8.00–6.84 (m, 9H, Harom), 6.08 (d, J = 7.9 Hz, 1H, NH), 5.30–5.22 (m, 3H, H-3’, H-4’, H-2”), 5.19 (d, J = 3.6 Hz, 1H, H-1’), 4.87 (ddd, J = 2.4, 10.3 Hz, JH,F = 27.1 Hz, 1H, H-3”), 4.79–4.53 (m, 7H, H-1, H-6ab, H-1”, H-4”, CH2Ph), 4.47 (m, 1H, H5’), 4.36–4.33 (m, 2H, H-6ab”), 4.14 (t, J = 6.2 Hz, 1H, H-3), 3.97 (t, J = 6.1 Hz, 1H, H-4), 3.90–3.83 (m, 2H, H-5, H-2’), 3.76 (s, 3H, OCH3), 3.69–3.61 (m, 2H, H-2, H-5”), 3.34 (s, 3H, OCH3), 2.11, 1.95, 1.87 (3s, 9H, 3 × C(O)CH3), 1.17–1.14 (m, 30H, H-6’, 3 × C(CH3)3); 13C NMR (100 MHz, CDCl3, 295 K) δ 177.6, 177.0, 170.5, 170.4, 169.8, 166.0, 159.2 (C=O), 133.3, 130.3, 129.7, 129.5–128.6, 113.8 (Ar), 100.5 (C-1’), 99.2 (C-1”), 95.9 (C-1), 85.4 (d, JC,F = 186.3 Hz, C-4”), 73.3 (C-2’), 72.9 (C-4), 72.6 (CH2Ph), 72.1 (C-3), 71.6 (C-5), 71.1 (d, JC,F = 18.1 Hz, C-5”), 70.9 (d, JC,F = 18.5 Hz , C-3”), 70.5 (C-3’, C-4’), 68.5 (C-2”), 64.9 (C-5’), 63.8 (C-6), 60.7 (C-6”), 56.6, 55.2 (OCH3), 52.7 (C-2), 38.9, 38.8, 38.7 (C(CH3)3), 23.1 (C(CH3)3), 20.9, 20.7 (C(O)CH3), 15.8 (C-6’); HRMS–ESI (m/z): [M + H]+ calcd for C55H77FNO21, 1106.4972; found, 1106.4956.
References
-
Fukushi, Y.; Hakomori, S.-i.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685.
Return to citation in text: [1] [2] [3] -
Fukushi, Y.; Hakomori, S.-i.; Shepard, T. J. Exp. Med. 1984, 160, 506–520. doi:10.1084/jem.160.2.506
Return to citation in text: [1] [2] [3] -
Fukushi, Y.; Kannagi, R.; Hakomori, S.-i.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717.
Return to citation in text: [1] [2] [3] -
Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-i.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632.
Return to citation in text: [1] [2] [3] -
Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1989, 49, 3662–3669.
Return to citation in text: [1] [2] [3] -
Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1990, 50, 1375–1380.
Return to citation in text: [1] [2] [3] [4] -
Kobata, A.; Ginsburg, V. J. Biol. Chem. 1969, 244, 5496–5502.
Return to citation in text: [1] -
Yang, H.-J.; Hakomori, S.-i. J. Biol. Chem. 1971, 246, 1192–1200.
Return to citation in text: [1] -
Zhang, S.; Zhang, H. S.; Cordon-Cardo, C.; Reuter, V. E.; Singhal, A. K.; Lloyd, K. O.; Livingston, P. O. Int. J. Cancer 1997, 73, 50–56. doi:10.1002/(SICI)1097-0215(19970926)73:1<50::AID-IJC9>3.0.CO;2-0
Return to citation in text: [1] -
Satoh, J.; Kim, S. U. J. Neurosci. Res. 1994, 37, 466–474. doi:10.1002/jnr.490370406
Return to citation in text: [1] -
Croce, M. V.; Isla-Larrain, M.; Rabassa, M. E.; Demichelis, S.; Golussi, A. G.; Crespo, M.; Lacunza, E.; Segal-Eiras, A. Pathol. Oncol. Res. 2007, 13, 130–138. doi:10.1007/BF02893488
Return to citation in text: [1] -
Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3
Return to citation in text: [1] [2] [3] [4] [5] -
Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 1653–1664. doi:10.1016/j.carres.2008.04.017
Return to citation in text: [1] [2] -
Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Thøgersen, H.; Lemieux, R. U.; Bock, K.; Meyer, B. Can. J. Chem. 1982, 60, 44–57. doi:10.1139/v82-009
Return to citation in text: [1] -
Imberty, A.; Mikros, E.; Koča, J.; Mollicone, R.; Oriol, R.; Pérez, S. Glycoconjugate J. 1995, 12, 331–349. doi:10.1007/BF00731336
Return to citation in text: [1] -
Reynolds, M.; Fuchs, A.; Lindhorst, T. K.; Pérez, S. Mol. Simul. 2008, 34, 447–460. doi:10.1080/08927020701713878
Return to citation in text: [1] -
Miller, K. E.; Mukhopadhyay, C.; Cagas, P.; Bush, C. A. Biochemistry 1992, 31, 6703–6709. doi:10.1021/bi00144a009
Return to citation in text: [1] -
Haselhorst, T.; Weimar, T.; Peters, T. J. Am. Chem. Soc. 2001, 123, 10705–10714. doi:10.1021/ja011156h
Return to citation in text: [1] -
Pérez, S.; Mouhous-Riou, N.; Nifant’ev, N. E.; Tsvetkov, Y. E.; Bachet, B.; Imberty, A. Glycobiology 1996, 6, 537–542. doi:10.1093/glycob/6.5.537
Return to citation in text: [1] -
Bundle, D. R. Pure Appl. Chem. 1989, 61, 1171–1180. doi:10.1351/pac198961071171
Return to citation in text: [1] -
Meikle, P. J.; Young, N. M.; Bundle, D. R. J. Immunol. Methods 1990, 132, 255–261. doi:10.1016/0022-1759(90)90037-V
Return to citation in text: [1] -
Reimer, K. B.; Gidney, M. A. J.; Bundle, D. R.; Pinto, B. M. Carbohydr. Res. 1992, 232, 131–142. doi:10.1016/S0008-6215(00)91000-0
Return to citation in text: [1] -
Bundle, D. R. Recognition of Carbohydrate Antigens by Antibody Binding Sites. In Bioorganic Chemistry: Carbohydrates; Hecht, S. M., Ed.; Oxford University Press: New York, 1999; pp 370–440.
Return to citation in text: [1] [2] -
Lemieux, R. U. Acc. Chem. Res. 1996, 29, 373–380. doi:10.1021/ar9600087
Return to citation in text: [1] [2] -
Woods, R. J. Glycoconjugate J. 1998, 15, 209–216. doi:10.1023/A:1006984709892
Return to citation in text: [1] [2] -
Clarke, C.; Woods, R. J.; Gluska, J.; Cooper, A.; Nutley, M. A.; Boons, G.-J. J. Am. Chem. Soc. 2001, 123, 12238–12247. doi:10.1021/ja004315q
Return to citation in text: [1] [2] -
Laederach, A.; Reilly, P. J. Proteins: Struct., Funct., Bioinf. 2005, 60, 591–597. doi:10.1002/prot.20545
Return to citation in text: [1] [2] -
Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6
Return to citation in text: [1] [2] [3] [4] [5] -
Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc., Perkin Trans. 1 1979, 314–318. doi:10.1039/P19790000314
Return to citation in text: [1] [2] [3] -
Hindsgaul, O.; Norberg, T.; Le Pendu, J.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2
Return to citation in text: [1] [2] [3] -
Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3
Return to citation in text: [1] [2] [3] -
Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y
Return to citation in text: [1] [2] [3] -
Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9
Return to citation in text: [1] [2] [3] -
Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091
Return to citation in text: [1] [2] [3] -
Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 118, 9239–9248. doi:10.1021/ja9608555
Return to citation in text: [1] [2] [3] -
Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7
Return to citation in text: [1] [2] [3] -
Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2
Return to citation in text: [1] [2] [3] -
Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4
Return to citation in text: [1] [2] [3] -
Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034
Return to citation in text: [1] [2] [3] -
Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006
Return to citation in text: [1] [2] [3] -
Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l
Return to citation in text: [1] [2] [3] -
Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2
Return to citation in text: [1] [2] [3] -
La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7
Return to citation in text: [1] [2] [3] -
Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u
Return to citation in text: [1] [2] [3] -
Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529
Return to citation in text: [1] [2] [3] -
Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2
Return to citation in text: [1] [2] -
Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3
Return to citation in text: [1] [2] -
Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4
Return to citation in text: [1] [2] -
Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 739–758. doi:10.1080/07328309808002349
Return to citation in text: [1] [2] -
de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9
Return to citation in text: [1] [2] -
de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066
Return to citation in text: [1] [2] -
Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8
Return to citation in text: [1] [2] -
Sato, K.-i.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347
Return to citation in text: [1] [2] -
Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4
Return to citation in text: [1] [2] -
Risbood, P. A.; Reed, L. A., III; Goodman, L. Carbohydr. Res. 1981, 88, 245–251. doi:10.1016/S0008-6215(00)85538-X
Return to citation in text: [1] [2] -
Lattrell, R.; Lohaus, G. Justus Liebigs Ann. Chem. 1974, 901–920. doi:10.1002/jlac.197419740606
Return to citation in text: [1] -
Albert, R.; Dax, K.; Link, R. W.; Stütz, A. E. Carbohydr. Res. 1983, 118, C5–C6. doi:10.1016/0008-6215(83)88062-8
Return to citation in text: [1] -
Dong, H.; Pei, Z.; Ramström, O. J. Org. Chem. 2006, 71, 3306–3309. doi:10.1021/jo052662i
Return to citation in text: [1] -
Arita, H.; Ueda, N.; Matsushima, Y. Bull. Chem. Soc. Jpn. 1972, 45, 567–569. doi:10.1246/bcsj.45.567
Return to citation in text: [1] -
Kasuya, M. C.; Ito, A.; Hatanaka, K. J. Fluorine Chem. 2007, 128, 562–565. doi:10.1016/j.jfluchem.2007.02.013
Return to citation in text: [1] -
Subotkowski, W.; Friedrich, D.; Weiberth, F. J. Carbohydr. Res. 2011, 346, 2323–2326. doi:10.1016/j.carres.2011.07.019
Return to citation in text: [1] -
Paulsen, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 155–173. doi:10.1002/anie.198201553
Return to citation in text: [1] -
Crich, D.; Dudkin, V. J. Am. Chem. Soc. 2001, 123, 6819–6825. doi:10.1021/ja010086b
Return to citation in text: [1] -
Liao, L.; Auzanneau, F.-I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x
Return to citation in text: [1] [2] -
Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p
Return to citation in text: [1] [2] [3] -
Wang, A.; Hendel, J.; Auzanneau, F.-I. Beilstein J. Org. Chem. 2010, 6, No. 17. doi:10.3762/bjoc.6.17
Return to citation in text: [1] [2] [3] -
Wang, A.; Auzanneau, F.-I. Carbohydr. Res. 2010, 345, 1216–1221. doi:10.1016/j.carres.2010.03.038
Return to citation in text: [1] [2] -
Liao, L.; Auzanneau, F.-I. J. Org. Chem. 2005, 70, 6265–6273. doi:10.1021/jo050707+
Return to citation in text: [1] [2] -
Cheng, A.; Hendel, J. L.; Colangelo, K.; Bonin, M.; Auzanneau, F.-I. J. Org. Chem. 2008, 73, 7574–7579. doi:10.1021/jo801117y
Return to citation in text: [1] [2] -
Liao, L.; Robertson, V.; Auzanneau, F.-I. Carbohydr. Res. 2005, 340, 2826–2832. doi:10.1016/j.carres.2005.09.025
Return to citation in text: [1] -
Ruttens, B.; Kováč, P. Carbohydr. Res. 2006, 341, 1077–1080. doi:10.1016/j.carres.2006.04.007
Return to citation in text: [1] -
Kwon, Y.-U.; Soucy, R. L.; Snyder, D. A.; Seeberger, P. H. Chem.–Eur. J. 2005, 11, 2493–2504. doi:10.1002/chem.200400934
Return to citation in text: [1] -
Hendel, J. L.; Auzanneau, F.-I. Eur. J. Org. Chem. 2011, 6864–6876. doi:10.1002/ejoc.201101342
Return to citation in text: [1] -
Armarego, W. L. F.; Chai, C. L. L., Eds. Purification of Laboratory Chemicals, 5th ed.; Elsevier, 2003.
Return to citation in text: [1]
| 57. | Risbood, P. A.; Reed, L. A., III; Goodman, L. Carbohydr. Res. 1981, 88, 245–251. doi:10.1016/S0008-6215(00)85538-X |
| 58. | Lattrell, R.; Lohaus, G. Justus Liebigs Ann. Chem. 1974, 901–920. doi:10.1002/jlac.197419740606 |
| 59. | Albert, R.; Dax, K.; Link, R. W.; Stütz, A. E. Carbohydr. Res. 1983, 118, C5–C6. doi:10.1016/0008-6215(83)88062-8 |
| 60. | Dong, H.; Pei, Z.; Ramström, O. J. Org. Chem. 2006, 71, 3306–3309. doi:10.1021/jo052662i |
| 61. | Arita, H.; Ueda, N.; Matsushima, Y. Bull. Chem. Soc. Jpn. 1972, 45, 567–569. doi:10.1246/bcsj.45.567 |
| 1. | Fukushi, Y.; Hakomori, S.-i.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685. |
| 2. | Fukushi, Y.; Hakomori, S.-i.; Shepard, T. J. Exp. Med. 1984, 160, 506–520. doi:10.1084/jem.160.2.506 |
| 3. | Fukushi, Y.; Kannagi, R.; Hakomori, S.-i.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717. |
| 4. | Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-i.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632. |
| 5. | Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1989, 49, 3662–3669. |
| 6. | Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1990, 50, 1375–1380. |
| 12. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3 |
| 13. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 1653–1664. doi:10.1016/j.carres.2008.04.017 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 70. | Liao, L.; Auzanneau, F.-I. J. Org. Chem. 2005, 70, 6265–6273. doi:10.1021/jo050707+ |
| 71. | Cheng, A.; Hendel, J. L.; Colangelo, K.; Bonin, M.; Auzanneau, F.-I. J. Org. Chem. 2008, 73, 7574–7579. doi:10.1021/jo801117y |
| 1. | Fukushi, Y.; Hakomori, S.-i.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685. |
| 2. | Fukushi, Y.; Hakomori, S.-i.; Shepard, T. J. Exp. Med. 1984, 160, 506–520. doi:10.1084/jem.160.2.506 |
| 3. | Fukushi, Y.; Kannagi, R.; Hakomori, S.-i.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717. |
| 4. | Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-i.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632. |
| 5. | Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1989, 49, 3662–3669. |
| 6. | Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1990, 50, 1375–1380. |
| 30. | Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6 |
| 66. | Liao, L.; Auzanneau, F.-I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x |
| 72. | Liao, L.; Robertson, V.; Auzanneau, F.-I. Carbohydr. Res. 2005, 340, 2826–2832. doi:10.1016/j.carres.2005.09.025 |
| 73. | Ruttens, B.; Kováč, P. Carbohydr. Res. 2006, 341, 1077–1080. doi:10.1016/j.carres.2006.04.007 |
| 1. | Fukushi, Y.; Hakomori, S.-i.; Nudelman, E.; Cochran, N. J. Biol. Chem. 1984, 259, 4681–4685. |
| 2. | Fukushi, Y.; Hakomori, S.-i.; Shepard, T. J. Exp. Med. 1984, 160, 506–520. doi:10.1084/jem.160.2.506 |
| 3. | Fukushi, Y.; Kannagi, R.; Hakomori, S.-i.; Shepard, T.; Kulander, B. G.; Singer, J. W. Cancer Res. 1985, 45, 3711–3717. |
| 4. | Itzkowitz, S. H.; Yuan, M.; Fukushi, Y.; Palekar, A.; Phelps, P. C.; Shamsuddin, A. M.; Trump, B. T.; Hakomori, S.-i.; Kim, Y. S. Cancer Res. 1986, 46, 2627–2632. |
| 5. | Nakasaki, H.; Mitomi, T.; Noto, T.; Ogoshi, K.; Hanaue, H.; Tanaka, Y.; Makuuchi, H.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1989, 49, 3662–3669. |
| 6. | Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1990, 50, 1375–1380. |
| 25. | Bundle, D. R. Recognition of Carbohydrate Antigens by Antibody Binding Sites. In Bioorganic Chemistry: Carbohydrates; Hecht, S. M., Ed.; Oxford University Press: New York, 1999; pp 370–440. |
| 26. | Lemieux, R. U. Acc. Chem. Res. 1996, 29, 373–380. doi:10.1021/ar9600087 |
| 27. | Woods, R. J. Glycoconjugate J. 1998, 15, 209–216. doi:10.1023/A:1006984709892 |
| 28. | Clarke, C.; Woods, R. J.; Gluska, J.; Cooper, A.; Nutley, M. A.; Boons, G.-J. J. Am. Chem. Soc. 2001, 123, 12238–12247. doi:10.1021/ja004315q |
| 29. | Laederach, A.; Reilly, P. J. Proteins: Struct., Funct., Bioinf. 2005, 60, 591–597. doi:10.1002/prot.20545 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 67. | Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p |
| 7. | Kobata, A.; Ginsburg, V. J. Biol. Chem. 1969, 244, 5496–5502. |
| 8. | Yang, H.-J.; Hakomori, S.-i. J. Biol. Chem. 1971, 246, 1192–1200. |
| 9. | Zhang, S.; Zhang, H. S.; Cordon-Cardo, C.; Reuter, V. E.; Singhal, A. K.; Lloyd, K. O.; Livingston, P. O. Int. J. Cancer 1997, 73, 50–56. doi:10.1002/(SICI)1097-0215(19970926)73:1<50::AID-IJC9>3.0.CO;2-0 |
| 10. | Satoh, J.; Kim, S. U. J. Neurosci. Res. 1994, 37, 466–474. doi:10.1002/jnr.490370406 |
| 11. | Croce, M. V.; Isla-Larrain, M.; Rabassa, M. E.; Demichelis, S.; Golussi, A. G.; Crespo, M.; Lacunza, E.; Segal-Eiras, A. Pathol. Oncol. Res. 2007, 13, 130–138. doi:10.1007/BF02893488 |
| 25. | Bundle, D. R. Recognition of Carbohydrate Antigens by Antibody Binding Sites. In Bioorganic Chemistry: Carbohydrates; Hecht, S. M., Ed.; Oxford University Press: New York, 1999; pp 370–440. |
| 26. | Lemieux, R. U. Acc. Chem. Res. 1996, 29, 373–380. doi:10.1021/ar9600087 |
| 27. | Woods, R. J. Glycoconjugate J. 1998, 15, 209–216. doi:10.1023/A:1006984709892 |
| 28. | Clarke, C.; Woods, R. J.; Gluska, J.; Cooper, A.; Nutley, M. A.; Boons, G.-J. J. Am. Chem. Soc. 2001, 123, 12238–12247. doi:10.1021/ja004315q |
| 29. | Laederach, A.; Reilly, P. J. Proteins: Struct., Funct., Bioinf. 2005, 60, 591–597. doi:10.1002/prot.20545 |
| 70. | Liao, L.; Auzanneau, F.-I. J. Org. Chem. 2005, 70, 6265–6273. doi:10.1021/jo050707+ |
| 71. | Cheng, A.; Hendel, J. L.; Colangelo, K.; Bonin, M.; Auzanneau, F.-I. J. Org. Chem. 2008, 73, 7574–7579. doi:10.1021/jo801117y |
| 12. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3 |
| 13. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 1653–1664. doi:10.1016/j.carres.2008.04.017 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 67. | Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p |
| 6. | Singhal, A. K.; Ørntoft, T. F.; Nudelman, E.; Nance, S.; Schibig, L.; Stroud, M. R.; Clausen, H.; Hakomori, S.-i. Cancer Res. 1990, 50, 1375–1380. |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 68. | Wang, A.; Hendel, J.; Auzanneau, F.-I. Beilstein J. Org. Chem. 2010, 6, No. 17. doi:10.3762/bjoc.6.17 |
| 69. | Wang, A.; Auzanneau, F.-I. Carbohydr. Res. 2010, 345, 1216–1221. doi:10.1016/j.carres.2010.03.038 |
| 16. | Thøgersen, H.; Lemieux, R. U.; Bock, K.; Meyer, B. Can. J. Chem. 1982, 60, 44–57. doi:10.1139/v82-009 |
| 17. | Imberty, A.; Mikros, E.; Koča, J.; Mollicone, R.; Oriol, R.; Pérez, S. Glycoconjugate J. 1995, 12, 331–349. doi:10.1007/BF00731336 |
| 18. | Reynolds, M.; Fuchs, A.; Lindhorst, T. K.; Pérez, S. Mol. Simul. 2008, 34, 447–460. doi:10.1080/08927020701713878 |
| 19. | Miller, K. E.; Mukhopadhyay, C.; Cagas, P.; Bush, C. A. Biochemistry 1992, 31, 6703–6709. doi:10.1021/bi00144a009 |
| 20. | Haselhorst, T.; Weimar, T.; Peters, T. J. Am. Chem. Soc. 2001, 123, 10705–10714. doi:10.1021/ja011156h |
| 21. | Pérez, S.; Mouhous-Riou, N.; Nifant’ev, N. E.; Tsvetkov, Y. E.; Bachet, B.; Imberty, A. Glycobiology 1996, 6, 537–542. doi:10.1093/glycob/6.5.537 |
| 62. | Kasuya, M. C.; Ito, A.; Hatanaka, K. J. Fluorine Chem. 2007, 128, 562–565. doi:10.1016/j.jfluchem.2007.02.013 |
| 63. | Subotkowski, W.; Friedrich, D.; Weiberth, F. J. Carbohydr. Res. 2011, 346, 2323–2326. doi:10.1016/j.carres.2011.07.019 |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 22. | Bundle, D. R. Pure Appl. Chem. 1989, 61, 1171–1180. doi:10.1351/pac198961071171 |
| 23. | Meikle, P. J.; Young, N. M.; Bundle, D. R. J. Immunol. Methods 1990, 132, 255–261. doi:10.1016/0022-1759(90)90037-V |
| 24. | Reimer, K. B.; Gidney, M. A. J.; Bundle, D. R.; Pinto, B. M. Carbohydr. Res. 1992, 232, 131–142. doi:10.1016/S0008-6215(00)91000-0 |
| 64. | Paulsen, H. Angew. Chem., Int. Ed. Engl. 1982, 21, 155–173. doi:10.1002/anie.198201553 |
| 65. | Crich, D.; Dudkin, V. J. Am. Chem. Soc. 2001, 123, 6819–6825. doi:10.1021/ja010086b |
| 66. | Liao, L.; Auzanneau, F.-I. Org. Lett. 2003, 5, 2607–2610. doi:10.1021/ol034669x |
| 30. | Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6 |
| 31. | Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc., Perkin Trans. 1 1979, 314–318. doi:10.1039/P19790000314 |
| 32. | Hindsgaul, O.; Norberg, T.; Le Pendu, J.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2 |
| 33. | Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3 |
| 34. | Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y |
| 35. | Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9 |
| 36. | Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091 |
| 37. | Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 118, 9239–9248. doi:10.1021/ja9608555 |
| 38. | Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7 |
| 39. | Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2 |
| 40. | Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4 |
| 41. | Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034 |
| 42. | Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006 |
| 43. | Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l |
| 44. | Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2 |
| 45. | La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7 |
| 46. | Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u |
| 47. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 30. | Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6 |
| 31. | Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc., Perkin Trans. 1 1979, 314–318. doi:10.1039/P19790000314 |
| 32. | Hindsgaul, O.; Norberg, T.; Le Pendu, J.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2 |
| 33. | Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3 |
| 34. | Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y |
| 35. | Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9 |
| 36. | Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091 |
| 37. | Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 118, 9239–9248. doi:10.1021/ja9608555 |
| 38. | Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7 |
| 39. | Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2 |
| 40. | Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4 |
| 41. | Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034 |
| 42. | Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006 |
| 43. | Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l |
| 44. | Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2 |
| 45. | La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7 |
| 46. | Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u |
| 47. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 48. | Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2 |
| 49. | Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3 |
| 50. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 51. | Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 739–758. doi:10.1080/07328309808002349 |
| 52. | de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9 |
| 53. | de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066 |
| 54. | Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8 |
| 55. | Sato, K.-i.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347 |
| 15. | Wang, J.-W.; Asnani, A.; Auzanneau, F.-I. Bioorg. Med. Chem. 2010, 18, 7174–7185. doi:10.1016/j.bmc.2010.08.040 |
| 68. | Wang, A.; Hendel, J.; Auzanneau, F.-I. Beilstein J. Org. Chem. 2010, 6, No. 17. doi:10.3762/bjoc.6.17 |
| 69. | Wang, A.; Auzanneau, F.-I. Carbohydr. Res. 2010, 345, 1216–1221. doi:10.1016/j.carres.2010.03.038 |
| 74. | Kwon, Y.-U.; Soucy, R. L.; Snyder, D. A.; Seeberger, P. H. Chem.–Eur. J. 2005, 11, 2493–2504. doi:10.1002/chem.200400934 |
| 75. | Hendel, J. L.; Auzanneau, F.-I. Eur. J. Org. Chem. 2011, 6864–6876. doi:10.1002/ejoc.201101342 |
| 30. | Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6 |
| 68. | Wang, A.; Hendel, J.; Auzanneau, F.-I. Beilstein J. Org. Chem. 2010, 6, No. 17. doi:10.3762/bjoc.6.17 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 67. | Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 57. | Risbood, P. A.; Reed, L. A., III; Goodman, L. Carbohydr. Res. 1981, 88, 245–251. doi:10.1016/S0008-6215(00)85538-X |
| 56. | Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4 |
| 12. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3 |
| 30. | Dong, D.; Gourmala, C.; Zhang, Y.; Zhang, Y. Sci. China: Chem. 2010, 53, 1963–1969. doi:10.1007/s11426-010-4060-6 |
| 31. | Jacquinet, J.-C.; Sinaÿ, P. J. Chem. Soc., Perkin Trans. 1 1979, 314–318. doi:10.1039/P19790000314 |
| 32. | Hindsgaul, O.; Norberg, T.; Le Pendu, J.; Lemieux, R. U. Carbohydr. Res. 1982, 109, 109–142. doi:10.1016/0008-6215(82)84034-2 |
| 33. | Sato, S.; Ito, Y.; Nukada, T.; Nakahara, Y.; Ogawa, T. Carbohydr. Res. 1987, 167, 197–210. doi:10.1016/0008-6215(87)80279-3 |
| 34. | Jain, R. K.; Matta, K. L. Carbohydr. Res. 1992, 226, 91–100. doi:10.1016/0008-6215(92)84057-Y |
| 35. | Numomura, S.; Iida, M.; Numata, M.; Sugimoto, M.; Ogawa, T. Carbohydr. Res. 1994, 263, C1–C6. doi:10.1016/0008-6215(94)00264-9 |
| 36. | Jain, R. K.; Vig, R.; Rampal, R.; Chandrasekaran, E. V.; Matta, K. L. J. Am. Chem. Soc. 1994, 116, 12123–12124. doi:10.1021/ja00105a091 |
| 37. | Yan, L.; Kahne, D. J. Am. Chem. Soc. 1996, 118, 9239–9248. doi:10.1021/ja9608555 |
| 38. | Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171. doi:10.1016/S0008-6215(98)00148-7 |
| 39. | Figueroa-Pérez, S.; Verez-Bencomo, V. Tetrahedron Lett. 1998, 39, 9143–9146. doi:10.1016/S0040-4039(98)02104-2 |
| 40. | Cao, S.; Gan, Z.; Roy, R. Carbohydr. Res. 1999, 318, 75–81. doi:10.1016/S0008-6215(99)00080-4 |
| 41. | Gan, Z.; Cao, S.; Wu, Q.; Roy, R. J. Carbohydr. Chem. 1999, 18, 755–773. doi:10.1080/07328309908544034 |
| 42. | Zhang, Y.-M.; Esnault, J.; Mallet, J.-M.; Sinaÿ, P. J. Carbohydr. Chem. 1999, 18, 419–427. doi:10.1080/07328309908544006 |
| 43. | Zhu, T.; Boons, G.-J. J. Am. Chem. Soc. 2000, 122, 10222–10223. doi:10.1021/ja001930l |
| 44. | Zhu, T.; Boons, G.-J. Chem.–Eur. J. 2001, 7, 2382–2389. doi:10.1002/1521-3765(20010601)7:11<2382::AID-CHEM23820>3.0.CO;2-2 |
| 45. | La Ferla, B.; Prosperi, D.; Lay, L.; Giovanni, R.; Panza, L. Carbohydr. Res. 2002, 337, 1333–1342. doi:10.1016/S0008-6215(02)00164-7 |
| 46. | Xia, J.; Alderfer, J. L.; Locke, R. D.; Piskorz, C. F.; Matta, K. L. J. Org. Chem. 2003, 68, 2752–2759. doi:10.1021/jo020698u |
| 47. | Mukherjee, D.; Sarkar, S. K.; Chattopadhyay, P.; Chowdhury, U. S. J. Carbohydr. Chem. 2005, 24, 251–259. doi:10.1081/CAR-200058529 |
| 12. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3 |
| 14. | Hendel, J. L.; Cheng, A.; Auzanneau, F.-I. Carbohydr. Res. 2008, 343, 2914–2923. doi:10.1016/j.carres.2008.08.025 |
| 12. | Asnani, A.; Auzanneau, F.-I. Carbohydr. Res. 2003, 338, 1045–1054. doi:10.1016/S0008-6215(03)00053-3 |
| 48. | Toepfer, A.; Schmidt, R. R. Tetrahedron Lett. 1992, 33, 5161–5164. doi:10.1016/S0040-4039(00)79122-2 |
| 49. | Hummel, G.; Schmidt, R. R. Tetrahedron Lett. 1997, 38, 1173–1176. doi:10.1016/S0040-4039(97)00006-3 |
| 50. | Kretzschmar, G.; Stahl, W. Tetrahedron 1998, 54, 6341–6358. doi:10.1016/S0040-4020(98)00294-4 |
| 51. | Manzoni, L.; Lay, L.; Schmidt, R. R. J. Carbohydr. Chem. 1998, 17, 739–758. doi:10.1080/07328309808002349 |
| 52. | de la Fuente, J. M.; Penadés, S. Tetrahedron: Asymmetry 2002, 13, 1879–1888. doi:10.1016/S0957-4166(02)00480-9 |
| 53. | de Paz, J.-L.; Ojeda, R.; Barrientos, A. G.; Penadés, S.; Martín-Lomas, M. Tetrahedron: Asymmetry 2005, 16, 149–158. doi:10.1016/j.tetasy.2004.11.066 |
| 76. | Armarego, W. L. F.; Chai, C. L. L., Eds. Purification of Laboratory Chemicals, 5th ed.; Elsevier, 2003. |
| 54. | Windmüller, R.; Schmidt, R. R. Tetrahedron Lett. 1994, 35, 7927–7930. doi:10.1016/0040-4039(94)80013-8 |
| 55. | Sato, K.-i.; Seki, H.; Yoshimoto, A.; Nanaumi, H.; Takai, Y.; Ishido, Y. J. Carbohydr. Chem. 1998, 17, 703–727. doi:10.1080/07328309808002347 |
| 56. | Lönn, H. Carbohydr. Res. 1985, 139, 105–113. doi:10.1016/0008-6215(85)90011-4 |
© 2012 Moore and Auzanneau; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)