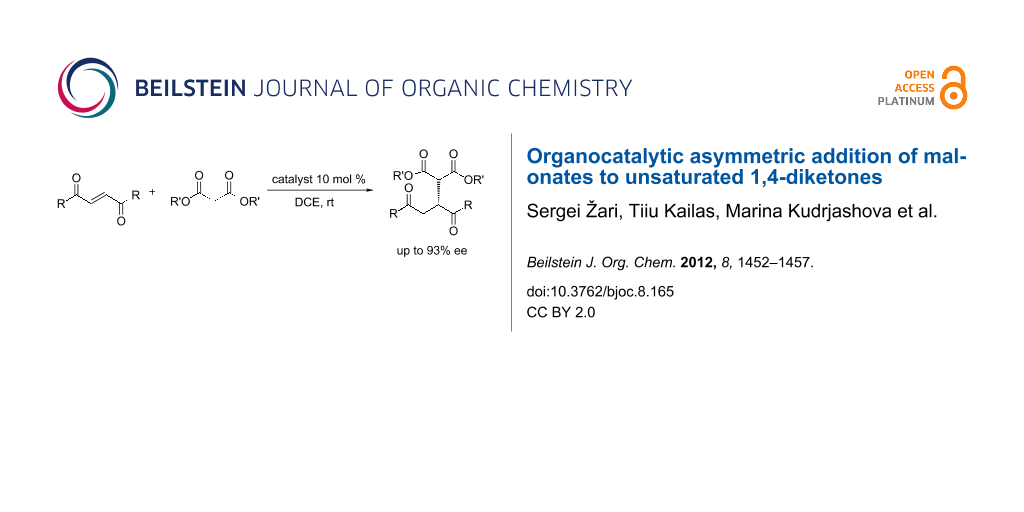
Abstract
The organocatalytic Michael addition of malonates to symmetric unsaturated 1,4-diketones catalyzed by thiourea and squaramide derivatives with Cinchona alkaloids afforded the formation of a new C–C bond in high yields (up to 98%) and enantiomeric purities (up to 93%). The absolute configuration of the product was suggested from comparison of the experimental and calculated VCD spectra of the reaction product 3a.

Graphical Abstract
Introduction
The asymmetric 1,4-conjugated addition (Michael reaction) of C-nucleophiles to enones is a powerful tool for obtaining a significant variety of enantioenriched products through a carbon–carbon bond formation [1-5]. Recently, unsaturated 1,4-dicarbonyl compounds, such as 1,4-ketoesters [6-8], 1,4-diketones [9], 1,4-ketoamides [9,10] and dialkylfumarates [11], have been the substrates for this reaction. The reaction products can undergo further chemical transformations, allowing the possibility of cascade reactions, making the method attractive for the synthesis of several valuable compounds, such as drugs and natural products. Tan et al. performed the addition of 1,3-alkylthiomalonates to 1,4-dicarbonylbut-2-enes, catalyzed by chiral bicyclic guanidines [7,9,12]. Xiao et al. reported the addition of nitroalkanes to 4-oxo-enoates, using chiral urea derivatives [7]. Miura et al. achieved an asymmetric addition of α,α-disubstituted aldehydes to maleimides catalyzed by primary amine thiourea organocatalyst [13]. Wang et al. reported the addition of dialkylmalonates and nitromethane to 4-oxo-4-arylbutenoates catalyzed by N,N´-dioxide-Sc(OTf)3 complexes [8]. Despite these and other successful experimental results, the asymmetric addition of malonates to symmetric aromatic unsaturated 1,4-diketones has not been systematically studied. Products of that reaction can be used as precursors of biologically active compounds. Padmaja et al. have reported that racemic heterocyclic compounds derived from the Michael addition of malonates and malononitrile to unsaturated 1,4-diketones possess antimicrobial and antifungal properties [14,15]. Therefore, new asymmetric additions of C-nucleophiles to unsaturated 1,4-diketones are highly in demand.
The asymmetric desymmetrization of symmetric unsaturated 1,4-diketones is a very challenging target. si-Attack on one carbon atom of the double bond and re-attack on the other leads to the same enantiomer. From the synthetic point of view, the conjugate addition of the nucleophile is, at the same time, a formal umpolung reaction with respect to the other carbonyl group (Figure 1).
![[1860-5397-8-165-1]](/bjoc/content/figures/1860-5397-8-165-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The conjugated addition to unsaturated 1,4-diketone 1.
Figure 1: The conjugated addition to unsaturated 1,4-diketone 1.
Results and Discussion
Catalyst screening
As a part of our ongoing studies in organocatalysis [16-19] we investigated the organocatalytic approach to the asymmetric desymmetrization of the title compounds with malonates. Three types of organocatalysts providing noncovalent interactions were used for this purpose: Cinchona alkaloids (I–V), thiourea derivatives (VI, VII) and squaramide derivatives (VIII, IX) (Figure 2). All of these screened catalysts are bifunctional compounds possessing hydrogen-bonding donor and acceptor moieties. Catalysts based on thiourea and squaramide differ from each other in their possible hydrogen-bond angles, rigidity of conformation, and pKa values [20]. Although the two squaramide based catalysts VIII and IX are structurally similar, they have quite different properties. Catalyst IX is a self-association-free compound [21], while catalyst VIII forms associates, and the stereoselectivity of the reaction in its presence depends on the catalyst concentration [22].
![[1860-5397-8-165-2]](/bjoc/content/figures/1860-5397-8-165-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The catalysts were screened in the reaction of phenyl disubstituted unsaturated 1,4-diketone 1a with diethyl malonate (2a, Table 1). The reaction was run in DCE at room temperature in the presence of 10 mol % of catalyst with a five-fold excess of malonate. In all cases, the yields of the products were very high. Cinchona alkaloids (Table 1, entries 1–4) catalyzed the reaction with low stereoselectivity. There was a remarkable difference in their reaction rates. Quinine (II) and quinidine (IV, Table 1, entries 2 and 4) were more efficient than cinchonine (I) and cinchonidine (III, Table 1, entries 1 and 3). The reduction of the vinyl group in quinine afforded dihydroquinine V. Unfortunately, no changes in the stereoselectivity of the model reaction were observed (Table 1, entry 5). Both thiourea catalysts derived from Cinchona alkaloids (VI, VII) gave high yields with good selectivities (Table 1, entries 6 and 7). Squaramide VIII and C2-symmetric squaramide IX gave good yields but slightly lower selectivities (Table 1, entries 8 and 9). The catalyst VII was selected for further studies as being the most efficient. Also, considering the partially aromatic character of the cyclobutenedione system, which may possibly allow additional interactions with the aromatic substrates 1, the catalyst IX was also chosen.
Table 1: Screening of the catalysts for the asymmetric conjugated-addition reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-165-i1.svg?max-width=637&scale=1.0)
|
|||||
| entry | catalyst | time (h) | yielda (%) | eeb (%) | abs. conf.c |
|---|---|---|---|---|---|
| 1 | I | 120 | 97 | 13 | R |
| 2 | II | 28 | 98 | 27 | R |
| 3 | III | 168 | 88 | 10 | R |
| 4 | IV | 24 | >98 | 19 | S |
| 5 | V | 20 | >98 | 18 | R |
| 6 | VI | 24 | >98 | 74 | S |
| 7 | VII | 19 | 98 | 74 | R |
| 8 | VIII | 15 | 96 | 60 | R |
| 9 | IX | 96 | 96 | 39 | R |
aIsolated yield; bdetermined by chiral HPLC; cdetermined by a comparison of the experimental and calculated VCD spectra (see the following).
Scope of the reaction
Next, we studied the effect of the malonate structure on the stereoselectivity of the reaction (Table 2). Although, the ester moiety can be replaced by other functional groups in the course of further synthetic transformations its main role is to provide the addition products with high ee value. The conditions for the reaction remained the same as they were in the catalyst screening experiments, except that a smaller excess of malonate (3 equiv, unless stated otherwise) was used. This did not influence the reaction time or the enantioselectivity, but afforded easier purification of the crude product.
Table 2: Enantioselective addition of malonates 2a-2f to unsaturated 1,4-diketone 1a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-165-i2.svg?max-width=637&scale=1.0)
|
|||||
| entry | R | catalyst | time (h) | yieldc (%) | eed (%) |
|---|---|---|---|---|---|
| 1 | a: Et | VII | 19 | 99 | 74 |
| 2 | a: Et | IX | 96 | 96 | 39 |
| 3 | b: Me | VII | 15 | 92 | 73 |
| 4 | b: Me | IX | 30 | 95 | 87 |
| 5 | c: iPr | VII | 36 | 96 | 69 |
| 6 | c: iPr | IX | 96 | 96 | 60 |
| 7a | d: t-Bu | VII | 75 | 70 | 59 |
| 8a | d: t-Bu | IX | 33 | 95 | 70 |
| 9b | e: Ph | VII | 3 | 44 | 37 |
| 10b | e: Ph | IX | 2 | 41 | 81 |
| 11 | f: Bn | VII | 6 | 95 | 68 |
| 12 | f: Bn | IX | 6 | 92 | 84 |
aReaction at 80 °C, malonate/dione 2:1; bmalonate/dione 1:1; cyields of isolated products; ddetermined by chiral HPLC.
1,4-Diketone 1a reacted smoothly with a variety of malonates 2a–2f, affording the products 3a–3f in high yields and with moderate to high stereoselectivities. In the case of the catalyst VII, the increase of steric hindrance of the malonate (Table 2, entries 1, 3, 5 and 7) led to a gradual drop in selectivity. Sterically more demanding malonates with branched alkyl or aryl groups (2c–e) gave products in much lower enantioselectivity (ee 37–69%) than the simple alkyl malonates (2a, b) (ee 73–74%). There was no clearly observed similar dependence with squaramide catalyst IX. Almost equally high ee values were obtained with methyl, phenyl or benzyl malonates (Table 2, entries 4, 10 and 12). A possible reason for the high selectivity with the phenyl-ring-containing esters could be the aromatic nature of the squaramide functional group in catalyst IX, allowing additional π–π-interactions.
The properties of the enone double bond of the substrate depend on the nature of the substituents in the phenyl ring. Therefore, the electronic effect of the para-substituent of unsaturated 1,4-diketone 1 on the reaction was investigated (Table 3). Electron-withdrawing groups, such as bromo and nitro, (Table 3, entries 10 and 13) as well as the electron-donating methoxy group (Table 3, entry 7) led to an increase in stereoselectivity, but the reaction time was also increased and the yields were lower with catalyst VII. A methyl substituent slightly decreased the enantioselectivity (Table 3, entry 4). This means that these dependencies cannot be clearly rationalized by the use of the electronic effects of the substituents in the phenyl ring. In the case of squaramide catalyst IX, all of the reactions became sluggish at room temperature (Table 3, entries 5, 8, 11 and 14). The reaction times were unreasonably long and the yields remained low. However, in the case of reactions with electron-withdrawing groups, the enantiomeric purity of the products was higher (Table 3, entries 11 and 14).
Table 3: Enantioselective addition of diethylmalonate 2a to substituted 1,4-diketones 1a, g–j.
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-165-i3.svg?max-width=637&scale=1.0)
|
||||||
| entry | R | catalyst | temp. (°C) | time (h) | yieldb (%) | eec (%) |
|---|---|---|---|---|---|---|
| 1 | a: H | VII | rt | 19 | 99 | 74 |
| 2 | a: H | IX | rt | 96 | 97 | 39 |
| 3 | a: H | IX | 80 | 10 | 97 | 82 |
| 4 | g: Me | VII | rt | 18 | 96 | 69 |
| 5 | g: Me | IX | rt | 213 | 25 | 18 |
| 6 | g: Me | IX | 80 | 22 | 76 | 66 |
| 7 | h: MeO | VII | rt | 88 | 90 | 79 |
| 8 | h: MeO | IX | rt | 48 | — | — |
| 9 | h: MeO | IX | 80 | 22 | 90 | 83 |
| 10a | i: Br | VII | rt | 48 | 83 | 81 |
| 11a | i: Br | IX | rt | 123 | 20 | 93 |
| 12a | i: Br | IX | 80 | 6 | 86 | 91 |
| 13 | j: NO2 | VII | rt | 54 | 66 | 81 |
| 14 | j: NO2 | IX | rt | 94 | 44 | 89 |
| 15 | j: NO2 | IX | 80 | 6 | 98 | 89 |
aDione/malonate 1.2:1; byields of isolated products; cdetermined by chiral HPLC.
Increasing the temperature had a drastic positive effect on the reactions performed in the presence of catalyst IX. It was found that by raising the temperature to 80 °C it was possible to significantly decrease the reaction time and increase yields up to 98% with almost no negative effect on the stereoselectivity (ee 66–89%, Table 3, entries 6, 9, 12 and 15). Moreover, the compounds 3a and 3g were obtained in much higher enantioselectivities, and the reaction with the unsaturated 1,4-diketone containing the electron-donating substituent 1h, which did not react at room temperature, also afforded the product in good yield and selectivity (Table 3, entry 9). As the squaramide-type catalyst IX is known to be self-association-free [21], the increase in enantioselectivity at higher temperatures can be attributed to the thermodynamic control of the conjugate addition. At the same time, the increase in temperature resulted in a small drop in stereoselectivity for the model reaction with catalyst VII.
The mechanism of the reaction is believed to be similar to that previously reported for 1,3-dicarbonyl compounds and acyl phosphonates [23]. Squaramide IX is a bifunctional catalyst that simultaneously coordinates electrophilic unsaturated 1,4-diketone via hydrogen bonding and activates the nucleophilic malonate via the tertiary amine of the quinuclidine moiety. Due to the symmetry of the substrate, there is no regioselectivity problem. A face selection is determined by the different access of the nucleophile to the tertiary amino group between the side chains of the catalysts. The re-face of the Michael acceptor is shielded by the flat quinoline unit and the si-attack of the malonate is preferred, affording R-selectivity (Figure 3).
![[1860-5397-8-165-3]](/bjoc/content/figures/1860-5397-8-165-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Determination of the absolute configuration
The absolute configuration of the product 3a was determined by a comparison of the experimental and calculated vibrational circular dichroism (VCD) spectra. DFT calculations (method B3PW91/6-311G**) of a series of conformers of compound 3a with R-configuration were performed. Calculations of harmonic vibrational frequencies were carried out for all favored conformers to verify their stability. The Boltzmann distribution of the Gibbs energy showed that one conformation out of six is dominant (84%). The experimental and calculated IR spectra match well in the range 1500–1800 cm−1 (both experimental and calculated spectra are normalized to 100% by using the highest peak from that range, Figure 4A).
![[1860-5397-8-165-4]](/bjoc/content/figures/1860-5397-8-165-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Calculated (red) and experimental (blue) IR (A) and VCD spectrum (B) of compound (R)-3a.
Figure 4: Calculated (red) and experimental (blue) IR (A) and VCD spectrum (B) of compound (R)-3a.
The most characteristic peaks of VCD spectra are in the same region (Figure 4B). The good agreement between calculated and experimental spectra directly allows for the assignment of the absolute configuration of 3a as the R-enantiomer.
Conclusion
We have developed a highly enantioselective method for the desymmetrization of aromatic unsaturated 1,4-diketones through organocatalytic reactions with malonates. The reaction is catalyzed by thiourea and squaramide derivatives with Cinchona alkaloids and affords products in very high yields (up to 99%) and in high enantioselectivities (up to 93%). This enantioselective 1,4-addition to unsaturated 1,4-diketones affords valuable intermediates for further synthetic transformations.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization and computational data. | ||
| Format: PDF | Size: 308.3 KB | Download |
References
-
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Sulzer-Mossé, S.; Alexakis, A. Chem. Commun. 2007, 3123–3135. doi:10.1039/B701216K
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Chem. Rev. 2005, 105, 933–972. doi:10.1021/cr040602r
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] -
Roca-Lopez, D.; Sadaba, D.; Delso, I.; Herrera, R. P.; Tejero, T.; Merino, P. Tetrahedron: Asymmetry 2010, 21, 2561–2601. doi:10.1016/j.tetasy.2010.11.001
Return to citation in text: [1] -
Ye, W.; Leow, D.; Goh, S. L. M.; Tan, C.-T.; Chian, C.-H.; Tan, C.-H. Tetrahedron Lett. 2006, 47, 1007–1010. doi:10.1016/j.tetlet.2005.11.133
Return to citation in text: [1] -
Lu, H. H.; Wang, X.-F.; Yao, C.; Zhang, J.; Wu, H.; Xiao, W.-J. Chem. Commun. 2009, 4251–4253. doi:10.1039/b905033g
Return to citation in text: [1] [2] [3] -
Wang, Z.; Chen, D.; Yang, Z.; Bai, S.; Liu, X.; Lin, L.; Feng, X. Chem.–Eur. J. 2010, 16, 10130–10136. doi:10.1002/chem.201001129
Return to citation in text: [1] [2] -
Jiang, Z.; Yang, Y.; Pan, Y.; Zhao, Y.; Liu, H.; Tan, C.-H. Chem.–Eur. J. 2009, 15, 4925–4930. doi:10.1002/chem.200802601
Return to citation in text: [1] [2] [3] -
Zigterman, J. L.; Woo, J. C. S.; Walker, S. D.; Tedrow, J. S.; Borths, C. J.; Bunel, E. E.; Faul, M. M. J. Org. Chem. 2007, 72, 8870–8876. doi:10.1021/jo701682c
Return to citation in text: [1] -
Shintani, R.; Ueyama, K.; Yamada, I.; Hayashi, T. Org. Lett. 2004, 6, 3425–3427. doi:10.1021/ol048421z
Return to citation in text: [1] -
Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S. L. M.; Leow, D.; Soh, Y.-T.; Tan, C.-H. Adv. Synth. Catal. 2007, 349, 2454–2458. doi:10.1002/adsc.200700326
Return to citation in text: [1] -
Miura, T.; Masuda, A.; Ina, M.; Nakashima, K.; Nishida, S.; Tada, N.; Itoh, A. Tetrahedron: Asymmetry 2011, 22, 1605–1609. doi:10.1016/j.tetasy.2011.09.006
Return to citation in text: [1] -
Padmaja, A.; Reddy, G. S.; Mohan, A. V. N.; Padmavathi, V. Chem. Pharm. Bull. 2008, 56, 647–653. doi:10.1248/cpb.56.647
Return to citation in text: [1] -
Padmaja, A.; Payani, T.; Reddy, G. D.; Padmavathi, V. Eur. J. Med. Chem. 2009, 44, 4557–4566. doi:10.1016/j.ejmech.2009.06.024
Return to citation in text: [1] -
Kanger, T.; Kriis, K.; Laars, M.; Kailas, T.; Müürisepp, A.-M.; Pehk, T.; Lopp, M. J. Org. Chem. 2007, 72, 5168–5173. doi:10.1021/jo070524i
Return to citation in text: [1] -
Laars, M.; Ausmees, K.; Uudsemaa, M.; Tamm, T.; Kanger, T.; Lopp, M. J. Org. Chem. 2009, 74, 3772–3775. doi:10.1021/jo900322h
Return to citation in text: [1] -
Noole, A.; Borissova, M.; Lopp, M.; Kanger, T. J. Org. Chem. 2011, 76, 1538–1545. doi:10.1021/jo200095e
Return to citation in text: [1] -
Noole, A.; Pehk, T.; Järving, I.; Lopp, M.; Kanger, T. Tetrahedron: Asymmetry 2012, 23, 188–198. doi:10.1016/j.tetasy.2012.01.021
Return to citation in text: [1] -
Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem.–Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694
Return to citation in text: [1] -
Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun. 2009, 7224–7226. doi:10.1039/B917882A
Return to citation in text: [1] [2] -
Rho, H. S.; Oh, S. H.; Lee, J. W.; Lee, J. Y.; Chin, J.; Song, C. E. Chem. Commun. 2008, 1208–1210. doi:10.1039/b719811f
Return to citation in text: [1] -
Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803
Return to citation in text: [1]
| 1. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 2. | Sulzer-Mossé, S.; Alexakis, A. Chem. Commun. 2007, 3123–3135. doi:10.1039/B701216K |
| 3. | Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Chem. Rev. 2005, 105, 933–972. doi:10.1021/cr040602r |
| 4. | Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 5. | Roca-Lopez, D.; Sadaba, D.; Delso, I.; Herrera, R. P.; Tejero, T.; Merino, P. Tetrahedron: Asymmetry 2010, 21, 2561–2601. doi:10.1016/j.tetasy.2010.11.001 |
| 11. | Shintani, R.; Ueyama, K.; Yamada, I.; Hayashi, T. Org. Lett. 2004, 6, 3425–3427. doi:10.1021/ol048421z |
| 21. | Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun. 2009, 7224–7226. doi:10.1039/B917882A |
| 9. | Jiang, Z.; Yang, Y.; Pan, Y.; Zhao, Y.; Liu, H.; Tan, C.-H. Chem.–Eur. J. 2009, 15, 4925–4930. doi:10.1002/chem.200802601 |
| 10. | Zigterman, J. L.; Woo, J. C. S.; Walker, S. D.; Tedrow, J. S.; Borths, C. J.; Bunel, E. E.; Faul, M. M. J. Org. Chem. 2007, 72, 8870–8876. doi:10.1021/jo701682c |
| 23. | Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803 |
| 9. | Jiang, Z.; Yang, Y.; Pan, Y.; Zhao, Y.; Liu, H.; Tan, C.-H. Chem.–Eur. J. 2009, 15, 4925–4930. doi:10.1002/chem.200802601 |
| 21. | Lee, J. W.; Ryu, T. H.; Oh, J. S.; Bae, H. Y.; Jang, H. B.; Song, C. E. Chem. Commun. 2009, 7224–7226. doi:10.1039/B917882A |
| 6. | Ye, W.; Leow, D.; Goh, S. L. M.; Tan, C.-T.; Chian, C.-H.; Tan, C.-H. Tetrahedron Lett. 2006, 47, 1007–1010. doi:10.1016/j.tetlet.2005.11.133 |
| 7. | Lu, H. H.; Wang, X.-F.; Yao, C.; Zhang, J.; Wu, H.; Xiao, W.-J. Chem. Commun. 2009, 4251–4253. doi:10.1039/b905033g |
| 8. | Wang, Z.; Chen, D.; Yang, Z.; Bai, S.; Liu, X.; Lin, L.; Feng, X. Chem.–Eur. J. 2010, 16, 10130–10136. doi:10.1002/chem.201001129 |
| 22. | Rho, H. S.; Oh, S. H.; Lee, J. W.; Lee, J. Y.; Chin, J.; Song, C. E. Chem. Commun. 2008, 1208–1210. doi:10.1039/b719811f |
| 8. | Wang, Z.; Chen, D.; Yang, Z.; Bai, S.; Liu, X.; Lin, L.; Feng, X. Chem.–Eur. J. 2010, 16, 10130–10136. doi:10.1002/chem.201001129 |
| 16. | Kanger, T.; Kriis, K.; Laars, M.; Kailas, T.; Müürisepp, A.-M.; Pehk, T.; Lopp, M. J. Org. Chem. 2007, 72, 5168–5173. doi:10.1021/jo070524i |
| 17. | Laars, M.; Ausmees, K.; Uudsemaa, M.; Tamm, T.; Kanger, T.; Lopp, M. J. Org. Chem. 2009, 74, 3772–3775. doi:10.1021/jo900322h |
| 18. | Noole, A.; Borissova, M.; Lopp, M.; Kanger, T. J. Org. Chem. 2011, 76, 1538–1545. doi:10.1021/jo200095e |
| 19. | Noole, A.; Pehk, T.; Järving, I.; Lopp, M.; Kanger, T. Tetrahedron: Asymmetry 2012, 23, 188–198. doi:10.1016/j.tetasy.2012.01.021 |
| 13. | Miura, T.; Masuda, A.; Ina, M.; Nakashima, K.; Nishida, S.; Tada, N.; Itoh, A. Tetrahedron: Asymmetry 2011, 22, 1605–1609. doi:10.1016/j.tetasy.2011.09.006 |
| 20. | Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K. A. Chem.–Eur. J. 2011, 17, 6890–6899. doi:10.1002/chem.201003694 |
| 7. | Lu, H. H.; Wang, X.-F.; Yao, C.; Zhang, J.; Wu, H.; Xiao, W.-J. Chem. Commun. 2009, 4251–4253. doi:10.1039/b905033g |
| 7. | Lu, H. H.; Wang, X.-F.; Yao, C.; Zhang, J.; Wu, H.; Xiao, W.-J. Chem. Commun. 2009, 4251–4253. doi:10.1039/b905033g |
| 9. | Jiang, Z.; Yang, Y.; Pan, Y.; Zhao, Y.; Liu, H.; Tan, C.-H. Chem.–Eur. J. 2009, 15, 4925–4930. doi:10.1002/chem.200802601 |
| 12. | Ye, W.; Jiang, Z.; Zhao, Y.; Goh, S. L. M.; Leow, D.; Soh, Y.-T.; Tan, C.-H. Adv. Synth. Catal. 2007, 349, 2454–2458. doi:10.1002/adsc.200700326 |
| 14. | Padmaja, A.; Reddy, G. S.; Mohan, A. V. N.; Padmavathi, V. Chem. Pharm. Bull. 2008, 56, 647–653. doi:10.1248/cpb.56.647 |
| 15. | Padmaja, A.; Payani, T.; Reddy, G. D.; Padmavathi, V. Eur. J. Med. Chem. 2009, 44, 4557–4566. doi:10.1016/j.ejmech.2009.06.024 |
© 2012 Žari et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)