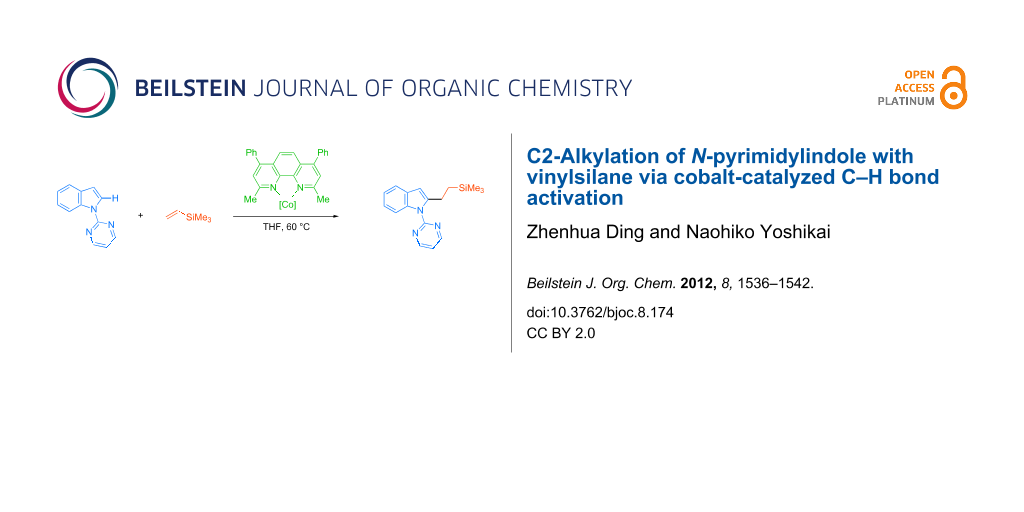
Abstract
Direct C2-alkylation of an indole bearing a readily removable N-pyrimidyl group with a vinylsilane was achieved by using a cobalt catalyst generated in situ from CoBr2, bathocuproine, and cyclohexylmagnesium bromide. The reaction allows coupling between a series of N-pyrimidylindoles and vinylsilanes at a mild reaction temperature of 60 °C, affording the corresponding alkylated indoles in moderate to good yields.

Graphical Abstract
Introduction
The indole ring ubiquitously occurs in biologically active natural and unnatural compounds [1-3]. Consequently, there has been a strong demand for catalytic methods allowing efficient and regioselective functionalization of indole derivatives [4-6]. Over the past decade, transition-metal-catalyzed direct functionalization has emerged as a powerful strategy for the direct introduction of aryl and alkenyl groups to the C2 and C3 positions of indole [7-9]. The situation is different when it comes to direct C–H alkylation [10,11]. The intrinsically nucleophilic C3 position of indole is amenable to a variety of catalytic alkylation reactions such as Friedel–Crafts reaction [5]. On the other hand, C2-alkylation of indoles has traditionally required 2-lithioindoles generated by C2-lithiation with a stoichiometric lithium base or indol-2-yl radicals generated from 2-halogenated indoles [12-17]. Examples of direct C2-alkylation via transition-metal-catalyzed C–H activation are still limited [18-20], while Jiao and Bach recently reported an elegant palladium-catalyzed, norbornene-mediated C2-alkylation reaction with a broad spectrum of alkyl bromides [21].
Over the past few years, our group and others have explored C–H bond functionalization reactions using cobalt complexes as inexpensive transition-metal catalysts [22], which often feature mild reaction conditions and unique regioselectivities [23-32]. As a part of this research program, we have recently reported a C2-alkenylation reaction of N-pyrimidylindoles with internal alkynes catalyzed by a cobalt–pyridylphosphine complex (Scheme 1a) [33], in which the pyrimidyl group functions as a readily removable directing group [34]. We also reported an ortho-alkylation reaction of aromatic imines with vinylsilanes and simple olefins using a cobalt–phenanthroline catalyst (Scheme 1b) [35]. Building on these studies, we have developed a cobalt–bathocuproine catalyst for the direct C2-alkylation reaction of N-pyrimidylindoles with vinylsilanes, which is reported herein (Scheme 1c).
![[1860-5397-8-174-i1]](/bjoc/content/inline/1860-5397-8-174-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) Cobalt-catalyzed C2-alkenylation of N-pyrimidylindole, (b) ortho-alkylation of aryl imine, and (c) C2-alkylation of N-pyrimidylindole.
Scheme 1: (a) Cobalt-catalyzed C2-alkenylation of N-pyrimidylindole, (b) ortho-alkylation of aryl imine, and ...
Results and Discussion
Our study commenced with the optimization of the reaction of N-pyrimidylindole 1a with vinyltrimethylsilane (2a). The combination of CoBr2 (10 mol %), 1,10-phenanthroline (phen, 10 mol %) and neopentylmagnesium bromide (100 mol %), which was effective for ortho-alkylation of aromatic imines [35], afforded the desired adduct 3aa in only 17% yield accompanied by a small amount of a C2-neopentylated product 4 (Table 1, entry 1). Subsequent examination of phenanthroline and bipyridine-type ligands (Table 1, entries 2–5) revealed that 2,9-dimethyl-1,10-phenanthroline (neocuproine) and 2,9-dimethyl-4,7-diphenylphenanthroline (bathocuproine) improved the yield of 3aa, while the byproduct 4 could not be suppressed (Table 1, entries 3 and 4). The P,N-bidentate ligand pyphos, which was the optimum ligand for the alkenylation reaction [33], was poorly effective (Table 1, entry 6). Additional screening of N-heterocyclic carbene (NHC) and phosphine ligands did not lead to an improvement of the catalytic efficiency (Table 1, entries 7–9). The reaction turned out to be sensitive to the amount of the Grignard reagent, as reduction of its loading from 100 to 60 mol % improved the yield of 3aa while suppressing the formation of byproduct 4 (Table 1, entry 10).
Table 1: Screening of ligands.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-174-i5.svg?max-width=637&scale=1.0)
|
|||
| entry | ligand (mol %) | yield (%)b | |
|---|---|---|---|
| 3aa | 4 | ||
| 1 | phen (10) | 17 | 7 |
| 2 | bathophen (10) | 11 | 7 |
| 3 | neocup (10) | 32 | 12 |
| 4 | bathocup (10) | 34 | 20 |
| 5 | dtbpy (10) | 1 | 3 |
| 6 | pyphos (10) | 2 | 3 |
| 7 | IMes·HCl (10) | 4 | 2 |
| 8 | PPh3 (20) | 9 | 5 |
| 9 | P(3-ClC6H4)3 (20) | 23 | 11 |
| 10c | bathocup | 50 | 10 |
aReaction was performed on a 0.3 mmol scale. bDetermined by GC using n-tridecane as an internal standard. c60 mol % of t-BuCH2MgBr was used.
Next, we performed screening of Grignard reagents using bathocuproine as the ligand (Table 2). Among Grignard reagents without β-hydrogen atoms, neopentyl- and phenylmagnesium bromides afforded 3aa in comparable yields (Table 2, entries 1 and 4), while trimethylsilylmethyl- and methylmagnesium chlorides gave much poorer results (Table 2, entries 2 and 3). Primary and secondary alkyl Grignard reagents also promoted the reaction, in which the reaction efficiency was strongly dependent on the alkyl group (Table 2, entries 5–10). We identified cyclohexylmagnesium bromide as the optimum Grignard reagent, which afforded 3aa in 69% isolated yield without formation of the cross-coupling product 4 between 1a and the Grignard reagent.
Table 2: Screening of Grignard reagents.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-174-i6.svg?max-width=637&scale=1.0)
|
|||
| entry | RMgX | yield (%)b | |
|---|---|---|---|
| 3aa | 4 | ||
| 1 | t-BuCH2MgBr | 50 | 10 |
| 2 | Me3SiCH2MgCl | 26 | 5 |
| 3 | MeMgCl | 14 | 4 |
| 4 | PhMgBr | 46 | 5 |
| 5 | EtMgBr | 28 | 3 |
| 6 | BuMgBr | 45 | 0 |
| 7 | i-PrMgBr | 49 | 3 |
| 8 | c-C3H5MgBr | 13 | 0 |
| 9 | c-C5H9MgBr | 46 | 0 |
| 10 | c-C6H11MgBr | 67 (69)c | 0 |
aReaction was performed on a 0.3 mmol scale. bDetermined by GC using n-tridecane as an internal standard. cIsolated yield.
With the optimized catalytic system in hand, we explored the scope of the reaction (Scheme 2). A variety of N-pyrimidylindoles participated in the reaction with vinyltrimethylsilane to afford the alkylation products 3ba–3ia in moderate yields, with tolerance of electron-withdrawing (F and Cl) and electron-donating (OMe) substituents and steric hindrance at the C3 and C7 positions. Unlike the cobalt-catalyzed C2-alkenylation reaction (Scheme 1a) [33], the reaction did not tolerate a cyano group on the indole substrate. In addition, N-pyrimidyl benzimidazole did not participate in the present alkylation reaction, although it was a good substrate for the C2-alkenylation reaction. A pyridyl group served as an alternative directing group to the pyrimidyl group, affording the alkylation product 3ka in 80% yield. On the other hand, an N,N-dimethylcarbamoyl group, which was previously used as a directing group for rhodium-catalyzed C2-alkenylation [36], was entirely ineffective. Vinylsilanes bearing dimethylphenylsilyl and triphenylsilyl groups were amenable to the addition reaction with 1a, affording the adduct 3ab and 3ac in modest yields. Vinyltriethoxysilane also reacted with 1a in 20% yield, although the product could not be separated in a pure form.
![[1860-5397-8-174-i2]](/bjoc/content/inline/1860-5397-8-174-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Addition of N-pyrimidylindoles to vinylsilanes.
Scheme 2: Addition of N-pyrimidylindoles to vinylsilanes.
Unfortunately, the present catalytic system was not very effective for C2-alkylation with simple olefins. The reaction of 1a with norbornene (2d) afforded the alkylation product 3ad in 30% yield (Scheme 3a). The reaction of 1-octene (2e) was even more sluggish, affording the alkylation product 3ae in only 9% yield (Scheme 3b). Styrene also reacted rather sluggishly to afford only a small amount of the alkylation product (3% as estimated by GC and GCMS), the regiochemistry (branched versus linear) of which has yet to be determined. An acrylate ester was not tolerable as an olefinic reaction partner because of the presence of excess Grignard reagent.
![[1860-5397-8-174-i3]](/bjoc/content/inline/1860-5397-8-174-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Addition of N-pyrimidylindole to norbornene (a) and 1-octene (b).
Scheme 3: Addition of N-pyrimidylindole to norbornene (a) and 1-octene (b).
The present alkylation reaction could be performed on a preparatively useful scale. Thus, alkylation of 1a with vinyltrimethylsilane (2a) on a 5 mmol scale afforded the adduct 3aa in 68% yield (Scheme 4). Furthermore, the pyrimidyl group on 3aa could be readily removed by heating with NaOEt in DMSO, affording the free indole 4aa in 85% yield.
![[1860-5397-8-174-i4]](/bjoc/content/inline/1860-5397-8-174-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gram-scale reaction and deprotection of N-pyrimidyl group.
Scheme 4: Gram-scale reaction and deprotection of N-pyrimidyl group.
Conclusion
In summary, we have developed a cobalt–bathocuproine catalyst for C2-alkylation of N-pyrimidyl indoles with vinylsilanes. The reaction could be performed at a mild temperature of 60 °C, on a preparatively useful scale. Ensuing studies will focus on the development of more broadly applicable catalytic systems for the direct alkylation of indole and other heterocycles.
Experimental
Typical procedure: Cobalt-catalyzed alkylation of N-pyrimidyl indole 1a with vinylsilane 2a
In a Schlenk tube were placed 1-(pyrimidin-2-yl)-1H-indole (1a) (58.6 mg, 0.3 mmol), CoBr2 (6.6 mg, 0.03 mmol), and bathocuproine (10.8 mg, 0.03 mmol), which were then dissolved in THF (1.3 mL). To the solution was added cyclohexylmagnesium bromide (0.60 M in THF, 0.3 mL, 0.18 mmol) at 0 °C. After stirring for 30 min at this temperature, vinyltrimethylsilane (2a) (66 μL, 0.45 mmol) was added. The reaction mixture was stirred at 60 °C for 12 h, and then quenched with saturated aqueous solution of NH4Cl (1.5 mL). The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was dried over Na2SO4 and concentrated under reduced pressure. Purification of the crude product by silica gel chromatography (eluent: hexane/EtOAc 100:1) afforded the title compound as a colorless oil (61.2 mg, 69%).
Supporting Information
| Supporting Information File 1: Experimental details and characterization data of new compounds. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Sundberg, R. J. Indoles; Academic Press: San Diego, CA, 1996.
Return to citation in text: [1] -
Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045–1075. doi:10.1039/A909834H
Return to citation in text: [1] -
Somei, M.; Yamada, F. Nat. Prod. Rep. 2005, 22, 73–103. doi:10.1039/b316241a
Return to citation in text: [1] -
Cacchi, S.; Fabrizi, G. Chem. Rev. 2011, 111, PR215–PR283. doi:10.1021/cr100403z
Return to citation in text: [1] -
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608–9644. doi:10.1002/anie.200901843
Return to citation in text: [1] [2] -
Seregin, I. V.; Gevorgyan, V. Chem. Soc. Rev. 2007, 36, 1173–1193. doi:10.1039/b606984n
Return to citation in text: [1] -
Joucla, L.; Djakovitch, L. Adv. Synth. Catal. 2009, 351, 673–714. doi:10.1002/adsc.200900059
Return to citation in text: [1] -
Boorman, T.; Larrosa, I. Recent advances in the C-2 regioselective direct arylation of indoles. In Progress in Heterocyclic Chemistry; Gribble, G. W.; Joule, J. A., Eds.; Elsevier: Oxford, U.K., 2010; Vol. 22, pp 1–20.
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131–4153. doi:10.1055/s-0030-1258262
Return to citation in text: [1] -
Ackermann, L. Chem. Commun. 2010, 46, 4866–4877. doi:10.1039/c0cc00778a
Return to citation in text: [1] -
Messaoudi, S.; Brion, J.-D.; Alami, M. Eur. J. Org. Chem. 2010, 6495–6516. doi:10.1002/ejoc.201000928
Return to citation in text: [1] -
Sundberg, R. J.; Russell, H. F. J. Org. Chem. 1973, 38, 3324–3330. doi:10.1021/jo00959a018
Return to citation in text: [1] -
Hasan, I.; Marinelli, E. R.; Lin, L.-C. C.; Fowler, F. W.; Levy, A. B. J. Org. Chem. 1981, 46, 157–164. doi:10.1021/jo00314a034
Return to citation in text: [1] -
Katritzky, A. R.; Akutagawa, K. Tetrahedron Lett. 1985, 26, 5935–5938. doi:10.1016/S0040-4039(00)98265-0
Return to citation in text: [1] -
Gharpure, M.; Stoller, A.; Bellamy, F.; Firnau, G.; Snieckus, V. Synthesis 1991, 1079–1082. doi:10.1055/s-1991-28395
Return to citation in text: [1] -
Fukuda, T.; Mine, Y.; Iwao, M. Tetrahedron 2001, 57, 975–979. doi:10.1016/S0040-4020(00)01063-2
Return to citation in text: [1] -
Fiumana, A.; Jones, K. Chem. Commun. 1999, 1761–1762. doi:10.1039/a905024h
Return to citation in text: [1] -
Nakao, Y.; Kashihara, N.; Kanyiva, K. S.; Hiyama, T. Angew. Chem., Int. Ed. 2010, 49, 4451–4454. doi:10.1002/anie.201001470
Return to citation in text: [1] -
Simon, M.-O.; Genet, J.-P.; Darses, S. Org. Lett. 2010, 12, 3038–3041. doi:10.1021/ol101038c
Return to citation in text: [1] -
Lee, D.-H.; Kwon, K.-H.; Yi, C. S. Science 2011, 333, 1613–1616. doi:10.1126/science.1208839
Return to citation in text: [1] -
Jiao, L.; Bach, T. J. Am. Chem. Soc. 2011, 133, 12990–12993. doi:10.1021/ja2055066
Return to citation in text: [1] -
Kulkarni, A. A.; Daugulis, O. Synthesis 2009, 4087–4109. doi:10.1055/s-0029-1217131
Return to citation in text: [1] -
Yoshikai, N. Synlett 2011, 1047–1051. doi:10.1055/s-0030-1259928
Return to citation in text: [1] -
Gao, K.; Lee, P.-S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2010, 132, 12249–12251. doi:10.1021/ja106814p
Return to citation in text: [1] -
Ding, Z.; Yoshikai, N. Org. Lett. 2010, 12, 4180–4183. doi:10.1021/ol101777x
Return to citation in text: [1] -
Gao, K.; Yoshikai, N. J. Am. Chem. Soc. 2011, 133, 400–402. doi:10.1021/ja108809u
Return to citation in text: [1] -
Lee, P.-S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2011, 133, 17283–17295. doi:10.1021/ja2047073
Return to citation in text: [1] -
Gao, K.; Yoshikai, N. Chem. Commun. 2012, 48, 4305–4307. doi:10.1039/c2cc31114c
Return to citation in text: [1] -
Chen, Q.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 428–429. doi:10.1021/ja1099853
Return to citation in text: [1] -
Ilies, L.; Chen, Q.; Zeng, X.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 5221–5223. doi:10.1021/ja200645w
Return to citation in text: [1] -
Li, B.; Wu, Z.-H.; Gu, Y.-F.; Sun, C.-L.; Wang, B.-Q.; Shi, Z.-J. Angew. Chem., Int. Ed. 2011, 50, 1109–1113. doi:10.1002/anie.201005394
Return to citation in text: [1] -
Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2012, 51, 775–779. doi:10.1002/anie.201106825
Return to citation in text: [1] -
Ding, Z.; Yoshikai, N. Angew. Chem., Int. Ed. 2012, 51, 4698–4701. doi:10.1002/anie.201200019
Return to citation in text: [1] [2] [3] -
Ackermann, L.; Lygin, A. V. Org. Lett. 2011, 13, 3332–3335. doi:10.1021/ol2010648
Return to citation in text: [1] -
Gao, K.; Yoshikai, N. Angew. Chem., Int. Ed. 2011, 50, 6888–6892. doi:10.1002/anie.201101823
Return to citation in text: [1] [2] -
Schipper, D. J.; Hutchinson, M.; Fagnou, K. J. Am. Chem. Soc. 2010, 132, 6910–6911. doi:10.1021/ja103080d
Return to citation in text: [1]
| 36. | Schipper, D. J.; Hutchinson, M.; Fagnou, K. J. Am. Chem. Soc. 2010, 132, 6910–6911. doi:10.1021/ja103080d |
| 1. | Sundberg, R. J. Indoles; Academic Press: San Diego, CA, 1996. |
| 2. | Gribble, G. W. J. Chem. Soc., Perkin Trans. 1 2000, 1045–1075. doi:10.1039/A909834H |
| 3. | Somei, M.; Yamada, F. Nat. Prod. Rep. 2005, 22, 73–103. doi:10.1039/b316241a |
| 5. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608–9644. doi:10.1002/anie.200901843 |
| 33. | Ding, Z.; Yoshikai, N. Angew. Chem., Int. Ed. 2012, 51, 4698–4701. doi:10.1002/anie.201200019 |
| 10. | Ackermann, L. Chem. Commun. 2010, 46, 4866–4877. doi:10.1039/c0cc00778a |
| 11. | Messaoudi, S.; Brion, J.-D.; Alami, M. Eur. J. Org. Chem. 2010, 6495–6516. doi:10.1002/ejoc.201000928 |
| 33. | Ding, Z.; Yoshikai, N. Angew. Chem., Int. Ed. 2012, 51, 4698–4701. doi:10.1002/anie.201200019 |
| 7. | Joucla, L.; Djakovitch, L. Adv. Synth. Catal. 2009, 351, 673–714. doi:10.1002/adsc.200900059 |
| 8. | Boorman, T.; Larrosa, I. Recent advances in the C-2 regioselective direct arylation of indoles. In Progress in Heterocyclic Chemistry; Gribble, G. W.; Joule, J. A., Eds.; Elsevier: Oxford, U.K., 2010; Vol. 22, pp 1–20. |
| 9. | Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131–4153. doi:10.1055/s-0030-1258262 |
| 35. | Gao, K.; Yoshikai, N. Angew. Chem., Int. Ed. 2011, 50, 6888–6892. doi:10.1002/anie.201101823 |
| 4. | Cacchi, S.; Fabrizi, G. Chem. Rev. 2011, 111, PR215–PR283. doi:10.1021/cr100403z |
| 5. | Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9608–9644. doi:10.1002/anie.200901843 |
| 6. | Seregin, I. V.; Gevorgyan, V. Chem. Soc. Rev. 2007, 36, 1173–1193. doi:10.1039/b606984n |
| 35. | Gao, K.; Yoshikai, N. Angew. Chem., Int. Ed. 2011, 50, 6888–6892. doi:10.1002/anie.201101823 |
| 22. | Kulkarni, A. A.; Daugulis, O. Synthesis 2009, 4087–4109. doi:10.1055/s-0029-1217131 |
| 33. | Ding, Z.; Yoshikai, N. Angew. Chem., Int. Ed. 2012, 51, 4698–4701. doi:10.1002/anie.201200019 |
| 21. | Jiao, L.; Bach, T. J. Am. Chem. Soc. 2011, 133, 12990–12993. doi:10.1021/ja2055066 |
| 34. | Ackermann, L.; Lygin, A. V. Org. Lett. 2011, 13, 3332–3335. doi:10.1021/ol2010648 |
| 18. | Nakao, Y.; Kashihara, N.; Kanyiva, K. S.; Hiyama, T. Angew. Chem., Int. Ed. 2010, 49, 4451–4454. doi:10.1002/anie.201001470 |
| 19. | Simon, M.-O.; Genet, J.-P.; Darses, S. Org. Lett. 2010, 12, 3038–3041. doi:10.1021/ol101038c |
| 20. | Lee, D.-H.; Kwon, K.-H.; Yi, C. S. Science 2011, 333, 1613–1616. doi:10.1126/science.1208839 |
| 12. | Sundberg, R. J.; Russell, H. F. J. Org. Chem. 1973, 38, 3324–3330. doi:10.1021/jo00959a018 |
| 13. | Hasan, I.; Marinelli, E. R.; Lin, L.-C. C.; Fowler, F. W.; Levy, A. B. J. Org. Chem. 1981, 46, 157–164. doi:10.1021/jo00314a034 |
| 14. | Katritzky, A. R.; Akutagawa, K. Tetrahedron Lett. 1985, 26, 5935–5938. doi:10.1016/S0040-4039(00)98265-0 |
| 15. | Gharpure, M.; Stoller, A.; Bellamy, F.; Firnau, G.; Snieckus, V. Synthesis 1991, 1079–1082. doi:10.1055/s-1991-28395 |
| 16. | Fukuda, T.; Mine, Y.; Iwao, M. Tetrahedron 2001, 57, 975–979. doi:10.1016/S0040-4020(00)01063-2 |
| 17. | Fiumana, A.; Jones, K. Chem. Commun. 1999, 1761–1762. doi:10.1039/a905024h |
| 23. | Yoshikai, N. Synlett 2011, 1047–1051. doi:10.1055/s-0030-1259928 |
| 24. | Gao, K.; Lee, P.-S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2010, 132, 12249–12251. doi:10.1021/ja106814p |
| 25. | Ding, Z.; Yoshikai, N. Org. Lett. 2010, 12, 4180–4183. doi:10.1021/ol101777x |
| 26. | Gao, K.; Yoshikai, N. J. Am. Chem. Soc. 2011, 133, 400–402. doi:10.1021/ja108809u |
| 27. | Lee, P.-S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2011, 133, 17283–17295. doi:10.1021/ja2047073 |
| 28. | Gao, K.; Yoshikai, N. Chem. Commun. 2012, 48, 4305–4307. doi:10.1039/c2cc31114c |
| 29. | Chen, Q.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 428–429. doi:10.1021/ja1099853 |
| 30. | Ilies, L.; Chen, Q.; Zeng, X.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 5221–5223. doi:10.1021/ja200645w |
| 31. | Li, B.; Wu, Z.-H.; Gu, Y.-F.; Sun, C.-L.; Wang, B.-Q.; Shi, Z.-J. Angew. Chem., Int. Ed. 2011, 50, 1109–1113. doi:10.1002/anie.201005394 |
| 32. | Yao, T.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2012, 51, 775–779. doi:10.1002/anie.201106825 |
© 2012 Ding and Yoshikai; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)