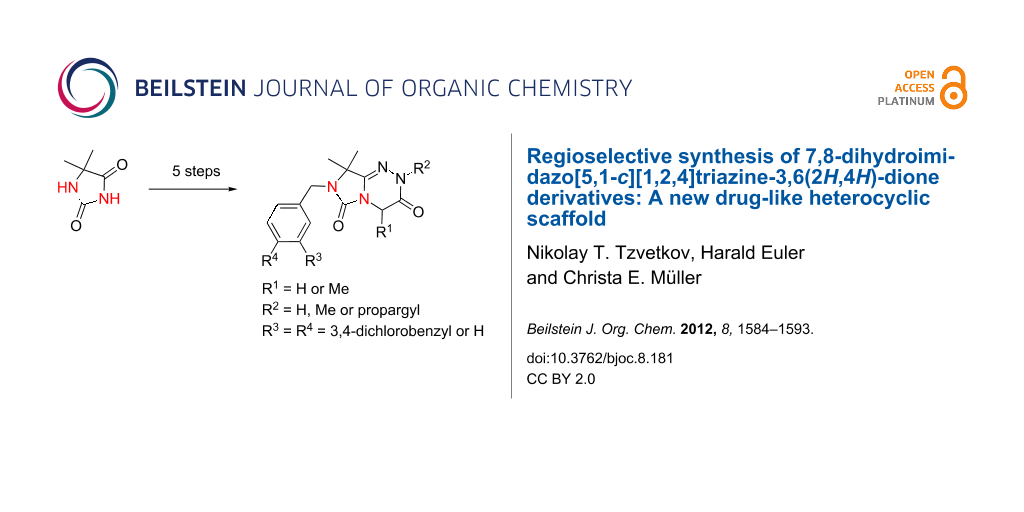
Abstract
Dihydroimidazo[5,1-c][1,2,4]triazine-3,6(2H,4H)-dione derivatives were prepared by successive N3- and N1-alkylation of hydantoins, followed by regioselective thionation and subsequent cyclization under mild conditions. In a final alkylation step a further substituent may be introduced. The synthetic strategy allows broad structural variation of this new drug-like heterobicyclic scaffold. In addition to extensive NMR and MS analyses, the structure of one derivative was confirmed by X-ray crystallography.

Graphical Abstract
Introduction
Imidazotriazines represent an important class of condensed heterobicycles that display a variety of significant biological activities, including anticancer [1], antimicrobial [2], anti-inflammatory [3] and neuroprotective [4] properties. The impressive array of biological effects of these compounds is associated with the 1,2,4-triazine ring as the core structural moiety, which also occurs in a number of natural products [5]. In addition, the 1,2,4-triazine scaffold has found application in pharmaceuticals and agrochemicals [6]. For example, some 7-phenylimidazo-[1,2.b][1,2,4]triazine derivatives of the general structure 1 (Figure 1) have been developed as selective ligands for γ-aminobutyric acid type A (GABAA) receptors and are therefore of benefit in the treatment and prevention of adverse conditions of the central nervous system (CNS), including anxiety, convulsions and cognitive disorders [7]. Novel fused 1,2,4-triazine-4(6H)-ones 2 showed selective cytotoxicity at micromolar concentrations against a wide range of cancer cells [1,8], while other derivatives of 2 exhibited analgesic [9], antibacterial, and antiviral activities [2]. Compounds with the 1,2,3,4-tetrahydroimidazo[1,2-b][1,2,4]triazine framework 3 were reported to be potentially useful as interleukin-1 (IL-1) and tumour necrosis factor (TNF) inhibitors for prophylactic and therapeutic treatment of chronic inflammatory diseases in which these cytokines are involved [3]. Recently, specifically substituted imidazo[1,5-f][1,2,4]triazines (4) have been developed as polo-like kinase (PLK) inhibitors with potential as anticancer therapeutics [10]. The 1,2,4-triazines 4 also exhibited inhibitory activities against glycogen synthase kinase 3 (GSK3β), and may therefore be developed for the treatment of haematological diseases, and inhibition of phosphodiesterase 10 (PDE10), which is potentially useful for the treatment of neurodegenerative diseases, especially Parkinson’s disease [11].
![[1860-5397-8-181-1]](/bjoc/content/figures/1860-5397-8-181-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active imidazo[1,2,4]triazine scaffolds 1–4.
Figure 1: Biologically active imidazo[1,2,4]triazine scaffolds 1–4.
The scaffolds 1–4 consist of a 1,2,4-triazine core, which arises from the ring fusion of C4a–N8 (1, 3 and 4) or C8a–N5 atoms (2), and differ in the arrangement of the substitution pattern of the imidazotriazine framework (Figure 1).
The structural variation of imidazo[1,2,4]triazine derivatives poses a significant challenge, particularly if a broad variety of substituents is to be introduced. Several synthetic strategies involving combinatorial and sequential approaches, in particular intramolecular cyclocondensation reactions of functionalized 1,2,4-triazole and imidazole precursors, have been developed [5,6,12].
We have been interested in expanding the medicinal-chemical space of synthetic drug-like small molecules focusing on 6,5-heterobicyclic ring systems in order to increase the diversity of our proprietary compound library [13]. Criteria for selection of the target structures include the potential for biological activity and bioavailability (peroral, and possibly central nervous system), structural novelty, synthetic accessibility, and the possibility for broad structural variations. In particular, we planned to introduce sp3-hybridized carbon atoms, to avoid completely flat aromatic structures that are prone to low solubility in water due to π-stacking effects.
We therefore identified the 1,2,4-triazine-containing scaffolds IV and VIII as promising novel target structures (Scheme 1). Depending on the starting material used, e.g., hydantoin I, or pyrazolidine-3,5-dione V, respectively, either 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-diones IV (route A) or 6,8-dihydropyrazolo[5,1-c][1,2,4]triazine-3,7-dione derivatives VIII (route B) should be accessible. In either case two nitrogen atoms, N7/N6 and N2, may be substituted with a variety of residues; e.g., alkylation of N2 (R5) in the very last step should allow an easy access to a library of compounds. Route B would proceed in four steps yielding product VIII starting from V, which may be obtained from the corresponding malonyl dichloride and hydrazine derivatives.
![[1860-5397-8-181-i1]](/bjoc/content/inline/1860-5397-8-181-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic approaches towards novel 7,8-dihydroimidazo-[5,1-c][1,2,4]-triazine-3,6-diones IV and 6,8-dihydropyrazolo[5,1-c][1,2,4]triazine-3,7-diones VIII.
Scheme 1: Retrosynthetic approaches towards novel 7,8-dihydroimidazo-[5,1-c][1,2,4]-triazine-3,6-diones IV an...
In the present study we focussed on synthetic route A starting from the commercially available hydantoins I, which would allow very broad structural diversity by introducing a variety of substituents and functionalities at different stages of the synthesis. As a first step ethyl (2,5-dioxoimidazolidin-1-yl)acetate derivative II is formed from hydantoins I. The condensation reaction of II with hydrazine, followed by a regioselective intramolecular heterocyclization, formally by dehydration, would afford the 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-diones III. The success of this route, however, depends on two factors: (i) the preferred regioselectivity of the successive N-alkylation steps for the formation of II; and (ii) the regioselective cyclization of 2,5-dioxoimidazolidines II to the desired product III, which is expected to be favoured in the case of dimethylhydantoin derivatives (R1 = R2 = Me).
Results and Discussion
Chemistry
Following the proposed strategy for the formation of 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-dione derivatives possessing aryl (R4 = 3,4-dichlorobenzyl) and alkyl/alkynyl (R5 = Me, propargyl) substituents at their nitrogen atoms 7 and 2, respectively, we started from the differently substituted hydantoins (R1 = R2 = H or Me) 5 and 6. N3- and subsequent N1-alkylation led to the N1,N3-dialkylated hydantoin derivatives 12–18. Alkylation reactions of hydantoins are well documented in the literature [14-18]. Depending on the alkylation reagents and reaction conditions, either N1- or N3-substituted hydantoins are accessible [14-17]. The details of the successfully conducted N3-alkylation of hydantoins 5 and 6 with various alkyl halides are shown in Table 1.
Table 1: Yields and reaction conditions for the N3-alkylation of hydantoin derivatives 5 and 6.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-181-i5.svg?max-width=637&scale=1.0)
|
||||
| starting compound | R2–X (1.1 equiv) | conditions | product | yield (%) |
|---|---|---|---|---|
| 5 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-181-i6.svg?max-width=637&scale=1.0)
|
1.1 equiv K2CO3
DMF, 85 °C, 42 h |
![[Graphic 3]](/bjoc/content/inline/1860-5397-8-181-i7.svg?max-width=637&scale=1.0)
7a |
89 |
| 6 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-8-181-i8.svg?max-width=637&scale=1.0)
|
1.1 equiv K2CO3
DMF, 80 °C, 56 h |
![[Graphic 5]](/bjoc/content/inline/1860-5397-8-181-i9.svg?max-width=637&scale=1.0)
8a |
76 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-8-181-i10.svg?max-width=637&scale=1.0)
|
1.1 equiv K2CO3
DMF, 85 °C, 90 h |
![[Graphic 7]](/bjoc/content/inline/1860-5397-8-181-i11.svg?max-width=637&scale=1.0)
9a |
92 |
| 6 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-8-181-i12.svg?max-width=637&scale=1.0)
= POM-Cl |
1.1 equiv K2CO3
DMF, 85 °C, 28 h |
![[Graphic 9]](/bjoc/content/inline/1860-5397-8-181-i13.svg?max-width=637&scale=1.0)
10 |
56 |
| 6 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-8-181-i14.svg?max-width=637&scale=1.0)
|
1.1 equiv K2CO3
DMF, 85 °C, 6 h |
![[Graphic 11]](/bjoc/content/inline/1860-5397-8-181-i15.svg?max-width=637&scale=1.0)
11b |
92 |
aCompounds 7 [19], 8 [19,20], 9 [19] have been described in the literature without detailed analytical data. bAnalytical data for 11 [14] are in accordance with literature data.
Alkylation of the thermodynamically preferred N3-position was achieved following a modified literature procedure, and the products could be obtained on a multigram scale [18]. In general, 1.1 equivalents of the corresponding alkylation reagent and potassium carbonate as a base in dry dimethylformamide (DMF) were used. The best yield (92%) was achieved with ethyl 2-bromopropionate and benzyl bromide as alkylating reagents. Although this procedure led to the direct formation of the desired N3-substituted products, the regioselectivity depended on the substitution pattern of 6, due to the directing effect of the methyl groups. Thus, traces of a N1-substituted regioisomer were detected by LC/ESI-MS analysis in the conversion of 5 to product 7 (N3/N1-alkylation ratio 15:1). Direct N1,N3-dialkylation of 5, however, was not observed. Alkylation with benzyl bromide was carried out in order to verify the N3-substitution yielding 11, which had already been described in the literature [14]. 3-Benzyl-5,5-dimethylhydantoin (11) was obtained in 92% isolated yield (see Section Experimental in Supporting Information File 1), in comparison to 80% obtained by the literature procedure [14]. Compounds 7–11 were isolated after chromatographic purification in good to excellent yields and analyzed by NMR spectroscopy (1H and 13C) and LC/ESI-MS. In addition, we obtained an X-ray crystal structure of 11 [18], which confirmed the alkylation at the N3-position of 5,5-dimethylhydantoin (6). The subsequent N1-alkylation of 2,5-dioxoimidazolidines 7–10 to the corresponding N,N-disubstituted hydantoins 12–18 was performed with 1.0–1.2 equiv of the appropriate alkylating reagent by using sodium hydride as a base in dry dimethylformamide (DMF) (Table 2). However, under these conditions the formation of compounds 12–18 strongly depended on the nature of the alkylating reagent, and therefore varying yields of dialkylated products were obtained. The reactions of 9 and 10 with 3,4-dichlorobenzyl bromide and phenethyl bromide, respectively, led to the formation of 15, 16, and 18 in poor to moderate yields. The alkylation with benzyl bromide worked well not only with sodium hydride (method 1) but also in the presence of potassium carbonate (method 2) as a base and gave rise to the disubstituted derivatives 12–14 and 17 in good yields. Products 12–18 were isolated after chromatographic purification (Table 2).
Table 2: Yields and reaction conditions of N1-alkylation of N3-substituted hydantoins 7–10.
![[Graphic 12]](/bjoc/content/inline/1860-5397-8-181-i16.svg?max-width=637&scale=1.0)
|
||||
| starting compound | R3–X (1.0–1.2 equiv) | conditions | product | yield (%) |
|---|---|---|---|---|
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-8-181-i17.svg?max-width=637&scale=1.0)
|
1.2 equiv NaH
DMF, 85 °C, 66 ha |
![[Graphic 14]](/bjoc/content/inline/1860-5397-8-181-i18.svg?max-width=637&scale=1.0)
12 |
70 |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-8-181-i19.svg?max-width=637&scale=1.0)
|
1.0 equiv NaH
DMF, 80 °C, 72 ha |
![[Graphic 16]](/bjoc/content/inline/1860-5397-8-181-i20.svg?max-width=637&scale=1.0)
13 |
80 |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-8-181-i21.svg?max-width=637&scale=1.0)
|
(1) 1.2 equiv NaHa
DMF, 85 °C, 80 h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-8-181-i22.svg?max-width=637&scale=1.0)
14 |
79a |
|
(2) 1.1 equiv K2CO3
DMF, 80 °C, 72 hb |
60b | |||
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-8-181-i23.svg?max-width=637&scale=1.0)
|
1.2 equiv NaH
DMF, 85 °C, 120 ha |
![[Graphic 20]](/bjoc/content/inline/1860-5397-8-181-i24.svg?max-width=637&scale=1.0)
15 |
37 |
| 9 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-8-181-i25.svg?max-width=637&scale=1.0)
|
1.1 equiv NaH
DMF, 85 °C, 68 ha |
![[Graphic 22]](/bjoc/content/inline/1860-5397-8-181-i26.svg?max-width=637&scale=1.0)
16 |
18 |
| 10 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-8-181-i27.svg?max-width=637&scale=1.0)
|
1.2 equiv K2CO3
DMF, 85 °C, 72 hb |
![[Graphic 24]](/bjoc/content/inline/1860-5397-8-181-i28.svg?max-width=637&scale=1.0)
17 |
62 |
| 10 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-8-181-i29.svg?max-width=637&scale=1.0)
|
1.2 equiv K2CO3
DMF, 85 °C, 69 hb |
![[Graphic 26]](/bjoc/content/inline/1860-5397-8-181-i30.svg?max-width=637&scale=1.0)
18 |
28 |
aMethod 1. bMethod 2.
In order to demonstrate the tractability of the successive N1,N3-alkylation of hydantoin 6 we applied a three-step synthetic route to obtain compound 19 [17]. N3-Unsubstituted, N1-substituted hydantoins were obtained by a three-step synthetic procedure by reaction of 6 with pivaloyloxymethyl chloride (POM-Cl) to introduce a protecting group prior to N1-alkylation. Subsequently, the ester can easily be cleaved with lithium hydroxide in a methanol/tetrahydrofuran mixture at room temperature to afford 1-benzyl-5,5-dimethylimidazolidine-2,4-dione (19) (Scheme 2).
![[1860-5397-8-181-i2]](/bjoc/content/inline/1860-5397-8-181-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of N3-unsubstituted, N1-substituted hydantoin 19 by using a protection strategy.
Scheme 2: Synthesis of N3-unsubstituted, N1-substituted hydantoin 19 by using a protection strategy.
According to our retrosynthetic analysis (Scheme 1), and based on an efficient protocol for the regioselective N-alkylation of hydantoins, we applied a four-step procedure for the synthesis of differently substituted 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-diones 23–29 (Scheme 3).
![[1860-5397-8-181-i3]](/bjoc/content/inline/1860-5397-8-181-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-diones 23–29. Reagents and conditions: (i) P2S5, dioxane, reflux, 24 h; (ii) hydrazine monohydrate (20 equiv), EtOH, reflux 5–10 h; (iii) MeI (for 26) or methyl methanesulfonate (for 27), NaH, DMF, r.t.; (iv) propargyl bromide (80% in toluene), NaH, 85 °C, 48 h.
Scheme 3: Synthesis of 7,8-dihydroimidazo[5,1-c][1,2,4]triazine-3,6-diones 23–29. Reagents and conditions: (i...
The main challenge in the synthesis of 23–25 was the construction of the 1,2,4-triazine ring. Direct reaction of 13–15 with hydrazine hydrate failed, and only the corresponding hydrazides were formed. Therefore, the C5 carbonyl group of 13–15 was regioselectively thionated with phosphorus pentasulfide in dioxane under reflux [21] (Scheme 4). The products were purified chromatographically and the preferred regioselectivity (C5 versus C2) was confirmed by NMR spectroscopy. The formation of C2-thiocarbonyl by-products was not observed under these reaction conditions. The highly regioselective formation of thioketones 20–22 may be due to electronic effects and prevention of the formation of certain tautomers by the C4-methyl groups in compounds 13–15 [22]. To investigate the influence of the C4-methyl groups on the regioselective thionation reaction, we treated unsubstituted 5,5-dimethylhydantoin 6 with phosphorus pentasulfide in dioxane for four hours. As reported in the literature, we observed only the preferred C4 thiocarbonyl product while the thionation of 5-unsubstituted hydantoin yields a mixture of 2- and 4-thionated products, indicating that the 5-dimethyl substitution was responsible for regioselective thionation [21].
The proposed intramolecular N1–C5 heterocondensation as a key step to form the desired imidazo[5,1-c][1,2,4]triazine-3,6-diones 23–25 was accomplished by reaction of the thioxoimidazolidines 20–22 with hydrazine hydrate [23-26]. Different reaction conditions were applied, e.g., variation of the solvent or the amount of hydrazine monohydrate that was used. The highest yields of compounds 23–25 (almost 90% on average) were obtained when the condensation was performed in ethanol with a large excess of hydrazine hydrate (20 equiv) under reflux for the appropriate reaction time. We observed that the adding of molecular sieves (4 Å) to the reaction medium greatly improved the yields of condensed products 23–25. The regioselective two-step cyclization of 14 yielding imidazo[5,1-c][1,2,4]triazine-3,6-dione (24, pathway A) via an N5–C5 ring fusion is outlined in Scheme 4. Precursors 13 and 15 follow the same pathway A. A dehydrothionated intermediate (e.g., 30 in the preparation of 24) is formed. Obviously, N1–C2 cyclization (pathway B) via 31 is not favoured, and 32 is not formed starting from 21. The products 23–25 were purified by column chromatography and obtained on a multigram scale. Finally, the imidazotriazines were further functionalized by an N-alkylation reaction using different alkylating reagents under basic conditions (Scheme 3). Alkylation of 24 with methyl iodide led to 26, whereas methyl methanesufonate was used as an alkylating reagent for the methylation of 25 yielding 27. Reaction of 24 and 25 with propargyl bromide (80% in toluene) under basic conditions at 85 °C yielded the N-propargyl derivatives 28 and 29 (Scheme 3). The final products 26–29 were purified by column chromatography followed by preparative RP-HPLC, or by recrystallization; the pure products were obtained in good yields.
![[1860-5397-8-181-i4]](/bjoc/content/inline/1860-5397-8-181-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed regioselective two-step cyclization pathway to form 24 from 14.
Scheme 4: Proposed regioselective two-step cyclization pathway to form 24 from 14.
Structural analyses
Regioselectivity of the thionation reaction of the C5-carbonyl group was an essential precondition for a successful heterocondensation step yielding the desired imidazo[5,1-c][1,2,4]triazine-3,6-dione derivatives 23–25. The structural assignment of the precursors (2,5-dioxoimidazolidines 13–15 and their thiocarbonyl analogues 20–22) reported herein is based on their spectral data and, if necessary, supported by MMFF94 force field conformational analytical data [27]. The most important 13C NMR chemical shifts δ (ppm) are reported in Table 3. In the case of C5-thionation, the 13C NMR signal for this carbon atom is shifted from about 176 to 208–209 ppm. In addition, the C4 signal is shifted from ca. 62 to 71 ppm upon thionation.
![[Graphic 27]](/bjoc/content/inline/1860-5397-8-181-i31.svg?max-width=637&scale=1.0)
The structure determination of the condensed key products 24 and 25 was carried out by heteronuclear correlation NMR (HSQC and HMBC) in combination with 1H and 13C NMR, and additionally by LC/ESI-MS (m/z 287 [M + H]+/285 [M − H]− for 24, 355 [M + H]+/353 [M − H]− for 25). Molecular modelling was performed to calculate the respective geometries by using the MMFF95 force field [27] assuming an N1–C5 ring fusion during the intramolecular condensation reaction yielding 24 and 25 (Figure 2).
![[1860-5397-8-181-2]](/bjoc/content/figures/1860-5397-8-181-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Optimized structure (MMFF95) and key HMBC correlations of imidazo[5,1-c][1,2,4]triazine-3,6(2H,4H)-diones 24 and 25.
Figure 2: Optimized structure (MMFF95) and key HMBC correlations of imidazo[5,1-c][1,2,4]triazine-3,6(2H,4H)-...
The structural assignments of 24 and 25 were confirmed by HSQC and, most importantly, by HMBC (see Supporting Information File 1). The analysis of two HMBC spectra of 24 and 25 showed key correlations between C8 and HC/HD as well as between C6 and HC/HD methylene protons. In the case of an alternative N1–C2 ring fusion (structure 32 in Scheme 4) such a correlation between the carbonyl C5-function and the methylene protons HC/HD would be not possible. The structure was additionally confirmed by X-ray analysis of 24 (Figure 3) [28]. The molecules in the crystal are held together by one type of intermolecular hydrogen bond, located between O2 and the hydrogen H1 of the N2, with a distance of d(O···H1–N2) = 1.9437(2) Å, d(H1–N2) = 0.93(2) Å and an angle of ![[Graphic 28]](/bjoc/content/inline/1860-5397-8-181-i32.svg?max-width=637&scale=1.18182) (O2–H1–N2) = 155(2)°.
(O2–H1–N2) = 155(2)°.
![[1860-5397-8-181-3]](/bjoc/content/figures/1860-5397-8-181-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagram of 24 showing the atomic numbering. The thermal ellipsoids are drawn at the 50% probability level.
Figure 3: ORTEP diagram of 24 showing the atomic numbering. The thermal ellipsoids are drawn at the 50% proba...
Physicochemical properties
In order to assess the physicochemical properties of imidazo[5,1-c][1,2,4]triazine-3,6-dione derivatives we determined water-solubility, log P, and pKa values for compound 25 as a representative of this new class of heterocyclic compounds. Water solubility at physiological pH of 7.4 was found to be 47 µg/mL, which is a suitable range for perorally active drugs. A pKa value of 10.0, and a logP value of 3 was determined. Thus, the molecule will be uncharged under physiological conditions and the logP value is in a range which allows us to predict oral bioavailability [29].
Conclusion
In conclusion, we have designed and synthesized novel fused 7,8-dihydroimidazo-[5,1-c][1,2,4]triazine-3,6(2H,4H)-dione derivatives, i.e., a novel class of small heterocyclic molecules with drug-like properties, thereby expanding the druggable chemical space. For this purpose, we have developed a four-step convergent synthetic concept to access the imidazo[5,1-c][1,2,4]triazine frameworks starting from the commercially available 5,5-dimethylhydantoin 6. The reaction sequence involves successive N-alkylations of the corresponding hydantoin, followed by C5-thionation and an intramolecular heterocondensation reaction with hydrazine hydrate as a key step affording the imidazo[5,1-c][1,2,4]triazine-3,6-dione derivatives 23–25 in a regioselective manner. The synthetic procedure was optimized for all steps and can easily be carried out on a multigram scale. The experimentally determined physicochemical properties of prototypic compound 25 are indicative of drug-like properties suitable for peroral application. Alkylation in the final step allows the introduction of additional diversity and the preparation of compound libraries. The new synthetic strategy should also allow for the preparation of other, related heterobicyclic systems possessing different ring-members, ring sizes, and a variety of substituents.
Supporting Information
| Supporting Information File 1: Assays for determination of physicochemical properties of 25, experimental details and copies of NMR (1D and 2D) and LC/ESI-MS spectra of compounds 24 and 25. | ||
| Format: PDF | Size: 1.5 MB | Download |
Acknowledgements
A. Reiner, S. Terhart-Krabbe, M. Schneider and H. Passgang are gratefully acknowledged for skillful technical assistance. Dr. G. Schnakenburg and C. Rödde, Department for X-ray Single Crystal Structure Solution, Institute for Inorganic Chemistry, are acknowledged for collecting the dataset of compound 24. Dr. F. Pineda and Dr. M. Wiese are acknowledged for the determination of physicochemical data of compound 25. We are grateful to the German Federal Ministry of Education and Research (BMBF) and UCB Pharma for financial support (BIOPHARMA Neuroallianz project).
References
-
Sztanke, K.; Pasternak, K.; Rzymowska, J.; Sztanke, M.; Kandefer-Szerszeń, M. Eur. J. Med. Chem. 2008, 43, 1085–1094. doi:10.1016/j.ejmech.2007.07.009
Return to citation in text: [1] [2] -
Sztanke, K.; Pasternak, K.; Rajtar, B.; Sztanke, M.; Majek, M.; Polz-Dacewicz, M. Bioorg. Med. Chem. 2007, 15, 5480–5486. doi:10.1016/j.bmc.2007.05.048
Return to citation in text: [1] [2] -
Oku, T.; Kawai, Y.; Marusawa, H.; Tanaka, H. Imidazotriazine derivatives. WO001992012154A1, July 23, 1992.
Return to citation in text: [1] [2] -
Siegel, S.; Wilmen, A.; Röhrig, S.; Svenstrup, N.; Gnoth, M. J.; Heitmeier, S.; Rester, U. Imidazo-, Pyrazolopyrazine and Imidazotriazine und ihre Verwendung. DE102007032349A1, Jan 15, 2009.
Return to citation in text: [1] -
Maechling, S.; Good, J.; Lindell, S. D. J. Comb. Chem. 2010, 12, 818–821. doi:10.1021/cc1001617
Return to citation in text: [1] [2] -
Kirkman, J. K.; Lindell, S. D.; Maechling, S.; Slawin, A. M. Z.; Moody, C. J. Org. Biomol. Chem. 2008, 6, 4452–4459. doi:10.1039/b810850a
Return to citation in text: [1] [2] -
Bettati, M.; Blurton, P.; Carling, W. R.; Chambers, M. S.; Halett, D. J.; Jennings, A.; Lewis, R. T.; Russell, M. G. N.; Street, L. J.; Szekeres, H. J.; Van Neil, M. B. Imidazo-triazine derivatives as ligands for GABA recaptors. US000006936608B2, Aug 30, 2005.
Return to citation in text: [1] -
Sztanke, K.; Tuzimski, T.; Sztanke, M.; Rzymowska, J.; Pasternak, K. Bioorg. Med. Chem. 2011, 19, 5103–5116. doi:10.1016/j.bmc.2011.07.027
Return to citation in text: [1] -
Sztanke, K.; Markowski, W.; Świeboda, R.; Polak, B. Eur. J. Med. Chem. 2010, 45, 2644–2649. doi:10.1016/j.ejmech.2010.01.068
Return to citation in text: [1] -
Cheung, M.; King, N. P.; Kuntz, K. W.; Mook, R. A., Jr.; Pobanz, M. A.; Salovich, J. M.; Wilson, B. J. Imidazotriazine compounds. US000007462614B2, Dec 9, 2008.
Return to citation in text: [1] -
Bauser, M.; Brückner, D.; Burkhardt, N.; Ergüden, J. K.; Flubacher, D.; Friedl, A.; Gerlach, I.; Hendrix, M.; Hinz, V.; Jork, R.; Naab, P.; Niewöhner, U.; Repp, T. O.; Schauss, D.; Schlemmer, K.-H.; Stoltefuss, J.; Tersteegen, A. Imidazotriazines for use as phosphodiesterase inhibitors. WO002003000693A1, Jan 30, 2003.
Return to citation in text: [1] -
Molina, P.; Lorenzo, A.; Aller, E. Synthesis 1989, 843–847. doi:10.1055/s-1989-27406
Return to citation in text: [1] -
Substanzbibliothek — Fachgruppe Pharmazie. http://mueller-group.pharma.uni-bonn.de/bibliothek (accessed May 4, 2012).
Return to citation in text: [1] -
Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518
Return to citation in text: [1] [2] [3] [4] [5] -
Orazi, O. O.; Corral, R. A. Tetrahedron 1961, 15, 93–99. doi:10.1016/0040-4020(61)80012-4
Return to citation in text: [1] [2] -
Katritzky, A. R.; Grzeskowiak, N. E.; Siddiqui, T.; Jayaram, C.; Vassilatos, S. N. J. Chem. Res., Synop. 1982, 528.
Return to citation in text: [1] [2] -
Schläpfer-Dähler, M.; Mukherjee-Müller, G.; Heimgartner, H. Helv. Chim. Acta 1992, 75, 1251–1261. doi:10.1002/hlca.19920750425
Return to citation in text: [1] [2] [3] -
Euler, H.; Barbier, B.; Tzvetkov, N. T.; Müller, C. E. Z. Kristallogr. - New Cryst. Struct. 2009, 224, 601–602. doi:10.1524/ncrs.2009.0264
Return to citation in text: [1] [2] [3] -
Haga, T.; Toki, T.; Koyanagi, T.; Asai, N.; Yoshida, K.; Imai, O.; Yamamoto, K. Organophosphorus based compound, productio thereof and insecticide, acaricide, nematicide and agent for killing insect pest in soil containing the same compound. JP000002000793A, Jan 5, 1990.
Return to citation in text: [1] [2] [3] -
Curtin, L. M.; Dai, Y.; Davidsen, S. K.; Dellaria, J. F., Jr.; Florjancic, A. S.; Gong, J.; Guo, Y.; Heyman, H. R.; Holms, J. H.; Michaelides, M. R.; Stacey, J. R.; Steinman, D. H.; Wada, C. K.; Xu, L. Reverse hydroxamate inhibitors of matrix metalloproteinases. US000006294573B1, Sept 25, 2001.
Return to citation in text: [1] -
Cristiani, F.; Devillanova, F. A.; Diaz, A.; Isaia, F.; Verani, G. Phosphorus Sulfur Relat. Elem. 1985, 22, 23–31. doi:10.1080/03086648508073350
Return to citation in text: [1] [2] -
Cherkasov, R. A.; Kutyrev, G. A.; Pudovik, A. N. Tetrahedron 1985, 41, 2567–2624. doi:10.1016/S0040-4020(01)96363-X
Return to citation in text: [1] -
Jakubkiene, V.; Paulauskaite, R.; Vainilavicius, P. Chem. Heterocycl. Compd. 2007, 43, 485–489. doi:10.1007/s10593-007-0070-5
Return to citation in text: [1] -
Eberle, M. K.; Schirm, P. J. Heterocycl. Chem. 1977, 14, 59–63. doi:10.1002/jhet.5570140111
Return to citation in text: [1] -
Brugger, M.; Wamhoff, H.; Korte, F. Justus Liebigs Ann. Chem. 1972, 755, 101–105. doi:10.1002/jlac.19727550112
Return to citation in text: [1] -
Deodhar, K. D.; D'Sa, A. D.; Pednekar, S. R.; Kanekar, D. S. Synthesis 1982, 853–854. doi:10.1055/s-1982-29972
Return to citation in text: [1] -
All calculations were carried out at a semiempirical level (AM1) with Titan V1.05, Schrödinger Inc., Wavefunction Inc. (18. August 2000).
Return to citation in text: [1] [2] -
CCDC–878004 (24) contains the supplementary crystallographic data for this paper. Copies of the data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (fax: +44-(0)1223-336033 or email: deposit@ccdc.cam.ac.uk).
Return to citation in text: [1] -
Keserü, G. M.; Makara, G. M. Nat. Rev. Drug Discovery 2009, 8, 203–212. doi:10.1038/nrd2796
Return to citation in text: [1]
| 22. | Cherkasov, R. A.; Kutyrev, G. A.; Pudovik, A. N. Tetrahedron 1985, 41, 2567–2624. doi:10.1016/S0040-4020(01)96363-X |
| 21. | Cristiani, F.; Devillanova, F. A.; Diaz, A.; Isaia, F.; Verani, G. Phosphorus Sulfur Relat. Elem. 1985, 22, 23–31. doi:10.1080/03086648508073350 |
| 23. | Jakubkiene, V.; Paulauskaite, R.; Vainilavicius, P. Chem. Heterocycl. Compd. 2007, 43, 485–489. doi:10.1007/s10593-007-0070-5 |
| 24. | Eberle, M. K.; Schirm, P. J. Heterocycl. Chem. 1977, 14, 59–63. doi:10.1002/jhet.5570140111 |
| 25. | Brugger, M.; Wamhoff, H.; Korte, F. Justus Liebigs Ann. Chem. 1972, 755, 101–105. doi:10.1002/jlac.19727550112 |
| 26. | Deodhar, K. D.; D'Sa, A. D.; Pednekar, S. R.; Kanekar, D. S. Synthesis 1982, 853–854. doi:10.1055/s-1982-29972 |
| 1. | Sztanke, K.; Pasternak, K.; Rzymowska, J.; Sztanke, M.; Kandefer-Szerszeń, M. Eur. J. Med. Chem. 2008, 43, 1085–1094. doi:10.1016/j.ejmech.2007.07.009 |
| 5. | Maechling, S.; Good, J.; Lindell, S. D. J. Comb. Chem. 2010, 12, 818–821. doi:10.1021/cc1001617 |
| 13. | Substanzbibliothek — Fachgruppe Pharmazie. http://mueller-group.pharma.uni-bonn.de/bibliothek (accessed May 4, 2012). |
| 4. | Siegel, S.; Wilmen, A.; Röhrig, S.; Svenstrup, N.; Gnoth, M. J.; Heitmeier, S.; Rester, U. Imidazo-, Pyrazolopyrazine and Imidazotriazine und ihre Verwendung. DE102007032349A1, Jan 15, 2009. |
| 14. | Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518 |
| 15. | Orazi, O. O.; Corral, R. A. Tetrahedron 1961, 15, 93–99. doi:10.1016/0040-4020(61)80012-4 |
| 16. | Katritzky, A. R.; Grzeskowiak, N. E.; Siddiqui, T.; Jayaram, C.; Vassilatos, S. N. J. Chem. Res., Synop. 1982, 528. |
| 17. | Schläpfer-Dähler, M.; Mukherjee-Müller, G.; Heimgartner, H. Helv. Chim. Acta 1992, 75, 1251–1261. doi:10.1002/hlca.19920750425 |
| 18. | Euler, H.; Barbier, B.; Tzvetkov, N. T.; Müller, C. E. Z. Kristallogr. - New Cryst. Struct. 2009, 224, 601–602. doi:10.1524/ncrs.2009.0264 |
| 3. | Oku, T.; Kawai, Y.; Marusawa, H.; Tanaka, H. Imidazotriazine derivatives. WO001992012154A1, July 23, 1992. |
| 11. | Bauser, M.; Brückner, D.; Burkhardt, N.; Ergüden, J. K.; Flubacher, D.; Friedl, A.; Gerlach, I.; Hendrix, M.; Hinz, V.; Jork, R.; Naab, P.; Niewöhner, U.; Repp, T. O.; Schauss, D.; Schlemmer, K.-H.; Stoltefuss, J.; Tersteegen, A. Imidazotriazines for use as phosphodiesterase inhibitors. WO002003000693A1, Jan 30, 2003. |
| 2. | Sztanke, K.; Pasternak, K.; Rajtar, B.; Sztanke, M.; Majek, M.; Polz-Dacewicz, M. Bioorg. Med. Chem. 2007, 15, 5480–5486. doi:10.1016/j.bmc.2007.05.048 |
| 5. | Maechling, S.; Good, J.; Lindell, S. D. J. Comb. Chem. 2010, 12, 818–821. doi:10.1021/cc1001617 |
| 6. | Kirkman, J. K.; Lindell, S. D.; Maechling, S.; Slawin, A. M. Z.; Moody, C. J. Org. Biomol. Chem. 2008, 6, 4452–4459. doi:10.1039/b810850a |
| 12. | Molina, P.; Lorenzo, A.; Aller, E. Synthesis 1989, 843–847. doi:10.1055/s-1989-27406 |
| 9. | Sztanke, K.; Markowski, W.; Świeboda, R.; Polak, B. Eur. J. Med. Chem. 2010, 45, 2644–2649. doi:10.1016/j.ejmech.2010.01.068 |
| 3. | Oku, T.; Kawai, Y.; Marusawa, H.; Tanaka, H. Imidazotriazine derivatives. WO001992012154A1, July 23, 1992. |
| 28. | CCDC–878004 (24) contains the supplementary crystallographic data for this paper. Copies of the data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (fax: +44-(0)1223-336033 or email: deposit@ccdc.cam.ac.uk). |
| 1. | Sztanke, K.; Pasternak, K.; Rzymowska, J.; Sztanke, M.; Kandefer-Szerszeń, M. Eur. J. Med. Chem. 2008, 43, 1085–1094. doi:10.1016/j.ejmech.2007.07.009 |
| 8. | Sztanke, K.; Tuzimski, T.; Sztanke, M.; Rzymowska, J.; Pasternak, K. Bioorg. Med. Chem. 2011, 19, 5103–5116. doi:10.1016/j.bmc.2011.07.027 |
| 10. | Cheung, M.; King, N. P.; Kuntz, K. W.; Mook, R. A., Jr.; Pobanz, M. A.; Salovich, J. M.; Wilson, B. J. Imidazotriazine compounds. US000007462614B2, Dec 9, 2008. |
| 29. | Keserü, G. M.; Makara, G. M. Nat. Rev. Drug Discovery 2009, 8, 203–212. doi:10.1038/nrd2796 |
| 7. | Bettati, M.; Blurton, P.; Carling, W. R.; Chambers, M. S.; Halett, D. J.; Jennings, A.; Lewis, R. T.; Russell, M. G. N.; Street, L. J.; Szekeres, H. J.; Van Neil, M. B. Imidazo-triazine derivatives as ligands for GABA recaptors. US000006936608B2, Aug 30, 2005. |
| 27. | All calculations were carried out at a semiempirical level (AM1) with Titan V1.05, Schrödinger Inc., Wavefunction Inc. (18. August 2000). |
| 6. | Kirkman, J. K.; Lindell, S. D.; Maechling, S.; Slawin, A. M. Z.; Moody, C. J. Org. Biomol. Chem. 2008, 6, 4452–4459. doi:10.1039/b810850a |
| 2. | Sztanke, K.; Pasternak, K.; Rajtar, B.; Sztanke, M.; Majek, M.; Polz-Dacewicz, M. Bioorg. Med. Chem. 2007, 15, 5480–5486. doi:10.1016/j.bmc.2007.05.048 |
| 27. | All calculations were carried out at a semiempirical level (AM1) with Titan V1.05, Schrödinger Inc., Wavefunction Inc. (18. August 2000). |
| 19. | Haga, T.; Toki, T.; Koyanagi, T.; Asai, N.; Yoshida, K.; Imai, O.; Yamamoto, K. Organophosphorus based compound, productio thereof and insecticide, acaricide, nematicide and agent for killing insect pest in soil containing the same compound. JP000002000793A, Jan 5, 1990. |
| 20. | Curtin, L. M.; Dai, Y.; Davidsen, S. K.; Dellaria, J. F., Jr.; Florjancic, A. S.; Gong, J.; Guo, Y.; Heyman, H. R.; Holms, J. H.; Michaelides, M. R.; Stacey, J. R.; Steinman, D. H.; Wada, C. K.; Xu, L. Reverse hydroxamate inhibitors of matrix metalloproteinases. US000006294573B1, Sept 25, 2001. |
| 14. | Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518 |
| 15. | Orazi, O. O.; Corral, R. A. Tetrahedron 1961, 15, 93–99. doi:10.1016/0040-4020(61)80012-4 |
| 16. | Katritzky, A. R.; Grzeskowiak, N. E.; Siddiqui, T.; Jayaram, C.; Vassilatos, S. N. J. Chem. Res., Synop. 1982, 528. |
| 17. | Schläpfer-Dähler, M.; Mukherjee-Müller, G.; Heimgartner, H. Helv. Chim. Acta 1992, 75, 1251–1261. doi:10.1002/hlca.19920750425 |
| 19. | Haga, T.; Toki, T.; Koyanagi, T.; Asai, N.; Yoshida, K.; Imai, O.; Yamamoto, K. Organophosphorus based compound, productio thereof and insecticide, acaricide, nematicide and agent for killing insect pest in soil containing the same compound. JP000002000793A, Jan 5, 1990. |
| 17. | Schläpfer-Dähler, M.; Mukherjee-Müller, G.; Heimgartner, H. Helv. Chim. Acta 1992, 75, 1251–1261. doi:10.1002/hlca.19920750425 |
| 21. | Cristiani, F.; Devillanova, F. A.; Diaz, A.; Isaia, F.; Verani, G. Phosphorus Sulfur Relat. Elem. 1985, 22, 23–31. doi:10.1080/03086648508073350 |
| 14. | Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518 |
| 18. | Euler, H.; Barbier, B.; Tzvetkov, N. T.; Müller, C. E. Z. Kristallogr. - New Cryst. Struct. 2009, 224, 601–602. doi:10.1524/ncrs.2009.0264 |
| 18. | Euler, H.; Barbier, B.; Tzvetkov, N. T.; Müller, C. E. Z. Kristallogr. - New Cryst. Struct. 2009, 224, 601–602. doi:10.1524/ncrs.2009.0264 |
| 14. | Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518 |
| 19. | Haga, T.; Toki, T.; Koyanagi, T.; Asai, N.; Yoshida, K.; Imai, O.; Yamamoto, K. Organophosphorus based compound, productio thereof and insecticide, acaricide, nematicide and agent for killing insect pest in soil containing the same compound. JP000002000793A, Jan 5, 1990. |
| 14. | Jordan, T. E.; Ginsburg, S. J. Am. Chem. Soc. 1949, 71, 2258. doi:10.1021/ja01174a518 |
© 2012 Tzvetkov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)