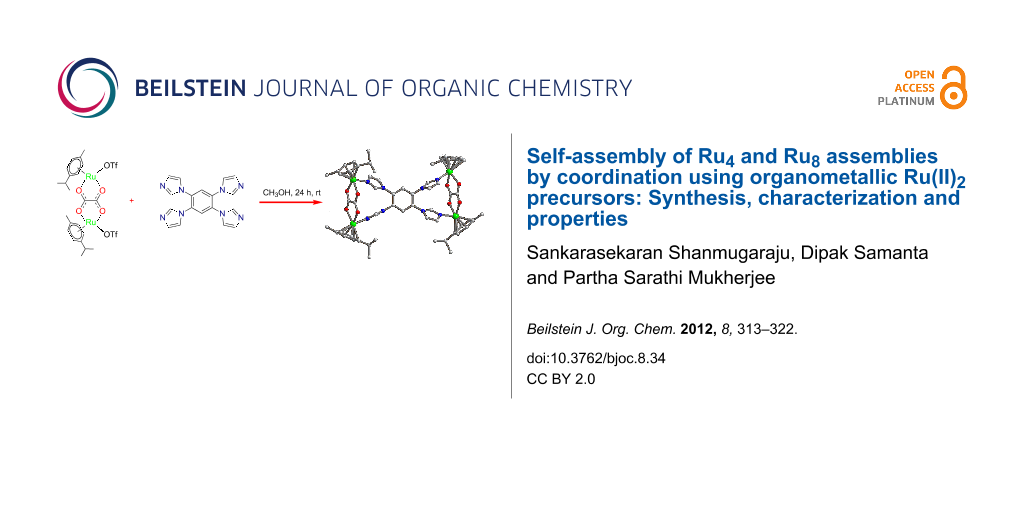
Abstract
Coordination-driven self-assembly of binuclear half-sandwich p-cymene ruthenium(II) complexes [Ru2(μ-η4-C2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1a) or [Ru2(μ-η4-N,N'-diphenyloxamidato)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1b) separately with an imidazole-based tetratopic donor L in methanol affords two tetranuclear metallamacrocycles 2a and 2b, respectively. Conversely, the similar combination of L with 2,5-dihydroxy-1,4-benzoquinonato (dhbq) bridged binuclear complex [Ru2(μ-η4-C6H2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1c) in 1:2 molar ratio resulted in an octanuclear macrocyclic cage 2c. All the self-assembled macrocycles 2a–2c were isolated as their triflate salts in high yields and were characterized fully by multinuclear (1H, 13C and 19F) NMR, infrared (IR) and electrospray ionization mass spectrometry (ESIMS). In addition, the molecular structure of macrocycle 2a was established unequivocally by single-crystal X-ray diffraction analysis and adopts a tetranuclear rectangular geometry with the dimensions of 5.53 Å × 12.39 Å. Furthermore, the photo- and electrochemical properties of these newly synthesized assemblies have been studied by using UV–vis absorption and cyclic voltammetry analysis.

Graphical Abstract
Introduction
Self-assembly of metal-based molecular architectures through coordination has emerged as an active field of research as large numbers of intricate structural motifs can easily be derived in a single step from predesigned molecular building units. In the last two decades several interesting molecular architectures of defined shapes, sizes and functionality have been discovered with the aid of this powerful protocol [1,2]. The basic requirement for successful metal–ligand-directed self-assembly resides in the judicious designing of information-encoding molecular building blocks having complementary binding sites. Large numbers of molecular polygons and polyhedra have been synthesized mostly from cis-blocked Pd(II)- and Pt(II)-based building units due to their rigid square-planar coordination geometry as well as to their interesting photophysical properties [3-5]. However, we and others have recently reported several shape-persistent discrete macrocycles/cages using half-sandwich octanuclear Ru(II) piano-stool complexes in combination with polypyridyl or polycarboxylate donors [6-10]. Ligands with an imidazole functionality are of considerable interest due to the presence of imidazole in many biological systems, and also the better donor character of imidazole compared to widely used pyridyl donors. Owing to these properties, we were interested in incorporating an imidazole functionality into discrete molecules of defined shapes, and also to investigate their influence in terms of directionality in controlling the geometry of the resulting molecular architectures. Herein, we report the self-assembly reactions of [Ru2(μ-η4-C2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1a) or [Ru2(μ-η4-N,N'-diphenyloxamidato)(MeOH)2(η6-p-cymene)2] (O3SCF3)2 (1b) and [Ru2(μ-η4-C6H2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1c) with a tetratopic imidazole-based donor L in methanol at room temperature to generate two tetranuclear macrocycles of general formula [Ru4(μ-η4-C2O4 or N,N'-diphenyloxamidato)2(L)(η6-p-cymene)4](O3SCF3)4 (2a, 2b) and an octanuclear macrocyclic cage [Ru8(μ-η4-C6H2O4)4(L)2(η6-p-cymene)8](O3SCF3)8 (2c), respectively in quantitative yields [L = 1,2,4,5-tetrakis(imidazol-1-yl)benzene]. The final assemblies were characterized by multinuclear (1H, 13C and 19F) NMR, IR and ESIMS analyses. The molecular structure of tetranuclear macrocycle 2a was determined by single-crystal X-ray diffraction analysis, which reveals a tetranuclear geometry with the dimensions of 5.53 Å × 12.39 Å. In addition to their synthesis and characterization, the UV–vis absorption and cyclic voltammetry studies are reported.
Results and Discussion
Synthesis and characterization of the tetranuclear complexes
According to the “directional-bonding approach” and “symmetry-interaction model”, one can expect the formation of either a [4 + 2] assembled octanuclear tetragonal prism or a [2 + 1] assembled tetranuclear 2D structure upon reaction of a binuclear “clip”-type acceptor and a tetratopic donor (Scheme 1) [11].
![[1860-5397-8-34-i1]](/bjoc/content/inline/1860-5397-8-34-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Possible octanuclear prism and tetranuclear macrocycle resulting from the combination of a tetradendate donor with a clip-type acceptor unit.
Scheme 1: Possible octanuclear prism and tetranuclear macrocycle resulting from the combination of a tetraden...
Instead, the exclusive formation of two tetranuclear assemblies (2a, 2b) upon mixing of binuclear Ru(II)2 acceptors 1a or 1b with an imidazole-based tetratopic donor L is interesting. The exclusive self-sorting of tetranuclear macrocycles (2a and 2b) rather than the expected octanuclear molecular cage is, presumably, due to the perfect matching of the distance between two Ru(II) metal centers with the distance between the adjacent imidazole donor sites in L. Moreover, from an entropic point of view such [2 + 1] self-assembly is expected to be preferred over [4 + 2] assembly as the loss of entropy in the latter case is more. As shown in Scheme 2, the binuclear acceptor 1a or 1b was treated separately with an imidazole-based tetratopic donor L in a 2:1 molar ratio in methanol at room temperature to obtain [2 + 1] self-assembled macrocycles 2a and 2b, respectively, in high yields. The addition of solid L into a methanolic solution of the acceptor (1a or 1b) lead to the immediate consumption of the solid and showed a sharp visible color change from light yellow to intense yellow. Both 2a and 2b are isolated as their triflate salts and are highly soluble in common organic solvents such as (CH3)2CO, CH3CN, CH3OH and CH2Cl2. The formation of the complexes (2a and 2b) was initially characterized by IR, multinuclear (1H, 13C and 19F) NMR and ESIMS spectrometric analyses.
![[1860-5397-8-34-i2]](/bjoc/content/inline/1860-5397-8-34-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of the tetranuclear complexes (2a and 2b) upon the reaction of Ru(II)2 based acceptors 1a or 1b and tetraimidazole ligand L in methanol at room temperature.
Scheme 2: Formation of the tetranuclear complexes (2a and 2b) upon the reaction of Ru(II)2 based acceptors 1a...
The IR spectra (Figure S1, Supporting Information File 1) of the macrocycles showed strong bands at 1624.6 cm−1 (2a) and 1602 cm−1 (2b) corresponding to the symmetrical stretching frequencies (νCO) of the carbonyl groups of the coordinated bis-bidentate oxalato (2a) or N,N'-diphenyloxamidato (2b) ligands. These bands, due to symmetrical stretching (νCO) in the complexes, are slightly shifted to a higher energy region compared to those of the starting acceptors (1a, νCO = 1623.7 cm−1; 1b, νCO = 1605.0 cm−1) due to the ligand-to-metal coordination. The appearance of a single peak in the 19F NMR spectrum at −81.03 ppm (2a) or −79.8 ppm (2b) indicated the presence of triflate counter anions in the same chemical environment in the resultant complexes (Figure 1 and Figure S3, Supporting Information File 1).
![[1860-5397-8-34-1]](/bjoc/content/figures/1860-5397-8-34-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H (left) and 19F (right) NMR spectra of tetranuclear macrocycle 2a recorded in CD2Cl2–CD3OD with peak assignments.
Figure 1: 1H (left) and 19F (right) NMR spectra of tetranuclear macrocycle 2a recorded in CD2Cl2–CD3OD with p...
Four peaks correspond to the donor L in the aromatic region (δ 6.95–8.10 ppm) were found in the 1H NMR spectra of 2a and 2b. Protons corresponding to the capped p-cymene moiety appeared in the range of δ 5.14–5.81 ppm. Notably, the 1H signals of the protons of L exhibited a significant downfield shift compared to the unbound linker due to the loss of electron density upon ligand-to-metal coordination (Figure 1 and Figure S2, Supporting Information File 1). The high solubility of the assemblies in common organic solvents and the symmetric nature of 1H NMR spectra primarily ruled out the formation of a polymer with extended coordination. Although the initial characterization of the self-assembled species by NMR spectroscopy intimated the ligand-to-metal coordination, it does not provide any information about the exact composition and nuclearity of the final products.
Electrospray ionization mass spectrometry (ESIMS) can be used as a soft-ionization method to elucidate the exact composition of the product [12]. The ESIMS analysis confirmed the formation of the rather unexpected [2 + 1] self-assembled tetranuclear macrocycles 2a and 2b by the appearance of multiply charged fragmented ions (Figure 2 and Figure S4, Supporting Information File 1). The multiply charged ions for 2a at m/z = 1906.9 [2a − O3SCF3−]+, 879.0 [2a − 2O3SCF3−]2+; for 2b at m/z = 1029.53 [2b − 2O3SCF3−]2+, 637.27 [2b − 3O3SCF3−]3+ were observed and these peaks were well resolved isotopically (Figure 2 and Figure S4, Supporting Information File 1). The appearance of the expected peaks along with the isotopic patterns was in strong support of the formation of [2 + 1] self-assembled products in both cases.
![[1860-5397-8-34-2]](/bjoc/content/figures/1860-5397-8-34-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ESIMS spectrum of the macrocycle 2a recorded in acetonitrile. Inset: Experimentally observed isotopic distribution for [2a − O3SCF3−]+ and [2a − 2O3SCF3−]2+ fragments.
Figure 2: ESIMS spectrum of the macrocycle 2a recorded in acetonitrile. Inset: Experimentally observed isotop...
Finally, the molecular structure of the tetranuclear metallamacrocycle [(p-cymene)4Ru4(μ-η4-oxalato)2(L)]4+(O3SCF3−)4 (2a) was unambiguously determined by single-crystal X-ray diffraction analysis. Single crystals of 2a of high enough quality for X-ray diffraction were grown by slow vapor diffusion of diethyl ether into a methanolic solution of 2a at room temperature. Macrocycle 2a was crystallized in a tetragonal crystal system with I41/a space group having sixteen formula units per unit cell. A perspective view of the macrocycle 2a is depicted in Figure 3 with atom numbering, and its selected bond parameters are summarized in Table S1, Supporting Information File 1.
![[1860-5397-8-34-3]](/bjoc/content/figures/1860-5397-8-34-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: A ball and stick representation of 2a with atom numbering. Color code: Ru = green; O = red; N = blue; C = dark gray. All hydrogen atoms, triflate counter anions, isopropyl and methyl groups of the p-cymene moiety are omitted for the sake of clarity.
Figure 3: A ball and stick representation of 2a with atom numbering. Color code: Ru = green; O = red; N = blu...
The crystal-structure analysis of 2a shows a tetranuclear geometry with dimensions of 5.53 Å × 12.39 Å, and the coordination geometry around each Ru(II) is octahedral. Capped p-cymene occupies three coordinating sites of each Ru(II). The bridging dianionic oxalato (C2O42−) ligand binds through its two oxygen atoms. The imidazole nitrogen of the donor L occupies the sixth coordination site. The oxalato ligand connects to two ruthenium metal centers through four oxygen atoms. The average Ru–N and Ru–O bond distances are 2.140 Å and 2.141 Å, respectively. The four donor sites of L are coordinated to four different Ru(II) metal centers and thereby it adopts a tetranuclear rectangular geometry. The solid-state packing of macrocycle 2a along the crystallographic c-axis results in a highly porous structure with square-type channels due to the π–π interactions between the adjacent macrocycles (Figure S8, Supporting Information File 1). Triflate (O3SCF3−) counter anions are located outside of the porous channel (Figure S8, Supporting Information File 1).
Synthesis and characterization of the octanuclear cage
The construction of an octanuclear macrocyclic cage 2c was accomplished following a similar procedure as adopted for the synthesis of tetranuclear rectangular complexes 2a and 2b. Drop-wise mixing of a methanolic solution of binuclear acceptor 1c into a suspension of tetradendate linker L in methanol in 2:1 molar ratio afforded the exclusive formation of the [4 + 2] self-assembled macrocyclic cage 2c in good yield after 24 h of stirring at room temperature (Scheme 3). The obvious reason for the formation of an octanuclear macrocyclic cage 2c in contrast to the tetranuclear rectangles 2a and 2b is attributed to the increased length of the acceptor unit 1c compared to 1a or 1b. The immediate consumption of the suspended donor L in the clear solution and the observable color change from purple to deep red indicated the progress of the self-assembly reaction. The initial characterization of the isolated macrocycle by multinuclear (1H, 13C and 19F) NMR suggested the formation of a single and symmetric macrocyclic complex.
![[1860-5397-8-34-i3]](/bjoc/content/inline/1860-5397-8-34-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of an octanuclear macrocycle 2c upon reaction of Ru(II)2-based acceptors 1c with imidazole-based tetratopic donor L in methanol at room temperature.
Scheme 3: Formation of an octanuclear macrocycle 2c upon reaction of Ru(II)2-based acceptors 1c with imidazol...
The infrared spectra (Figure S1, Supporting Information File 1) of the macrocycle (2c) showed a strong band at 1521.2 cm−1 corresponding to the symmetric stretching frequency (νCO) of the carbonyl groups of the coordinated bis-bidentate quinonato ligand, and this symmetric stretching frequency (νCO) in the isolated macrocycle is slightly shifted to the higher energy region compared to that of the starting acceptor (1c, νCO = 1515.8 cm−1) due to the ligand-to-metal coordination. The 1H NMR spectra of the macrocycle 2c established the expected peaks in the aromatic region corresponding to the tetratopic donor L and the proton resonance of capped p-cymene ligand. Moreover, the peak of L in the macrocyclic complex exhibits a significant downfield shift due to the ligand–metal coordination (Figure 4). The assignment of the proton signals in the 1H NMR spectra was confirmed by the 1H–1H COSY NMR (Figure S5, Supporting Information) spectral analysis. Furthermore, the appearance of prominent peaks in the ESIMS spectra of the multiply charged ions for 2c at m/z = 2008.0 [2c − 2O3SCF3−]2+, 1288.0 [2c − 3O3SCF3−]3+, 928.9 [2c − 4O3SCF3−]4+ indicated the formation of a [4 + 2] self-assembled octanuclear macrocyclic cage. The peaks corresponding to the [2c − 2O3SCF3−]2+ and [2c − 3O3SCF3−]3+ fragments are well resolved isotopically (Figure S6, Supporting Information File 1).
![[1860-5397-8-34-4]](/bjoc/content/figures/1860-5397-8-34-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H (left) and 19F (right) NMR spectrum of the macrocycle 2c recorded in CD3CN with the peak assignments.
Figure 4: 1H (left) and 19F (right) NMR spectrum of the macrocycle 2c recorded in CD3CN with the peak assignm...
Unfortunately, all efforts to obtain diffraction-quality single crystals of the macrocycle 2c have so far been unsuccessful. However, the analysis of multinuclear NMR (1H, 13C and 19F) in concurrence with the ESIMS study is fully consistent with the formation of a [4 + 2] self-assembled octanuclear metallacage. In view toward gaining further insight into the structural characteristics of the newly designed macrocycle, the energy-minimized structure of the macrocycle 2c was obtained by means of molecular mechanics universal-force-field simulation (MMUFF) [13]. A perspective view of the energy-minimized structure of the macrocycle 2c is depicted in Figure 5. The optimized structures of the macrocycle 2c suggested the formation of an octanuclear macrocycle having an internal diameter of 1.54 nm. Notably, the quinonato-bridged ruthenium clips are tilted out of the plane of the imidazole donor in 2c in order to minimize the steric influence between the acceptor clips. Therefore, macrocycle 2c adopts a staggered conformation as was observed in similar types of 3D cages [14].
![[1860-5397-8-34-5]](/bjoc/content/figures/1860-5397-8-34-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Side (left) and top (right) view of the energy-minimized structures of the octanuclear macrocycle 2c. Color code: green = Ru, blue = N, red = O, gray = C. The hydrogen atoms, methyl and isopropyl group of the cymene moiety have been removed for the sake of clarity.
Figure 5: Side (left) and top (right) view of the energy-minimized structures of the octanuclear macrocycle 2c...
UV–vis absorption and electrochemical studies
One of the strongest driving factors to design multifarious functional supramolecular complexes is attributed to their interesting functional properties. In general, the utilization of functional properties would be realized more from macrocycles containing transition metals, due to their high sensitivity towards various external stimuli, compared to purely organic macrocycles. In this regard, substantial efforts have been devoted to the design of novel, nanoscopic and functional metallamacrocycles and also to the study of their functional properties [15]. The photo- and electrochemical properties of our newly synthesized macrocycles (2a and 2b) are studied by UV–vis absorption and cyclic voltammetry investigations and the obtained results are summarized in Table 1.
Table 1: Results of UV–vis absorption (in CH3CN, 1.0 × 10−5 M) and electrochemical (in CH2Cl2, 0.1 M n-Bu4NPF6) studies of macrocycles 2a–2c at 298 K.
| Macrocycle |
Absorption maximaa
λmax (nm) |
Molar extinction coefficient
ε × 103 M−1cm−1 λmax (nm) |
E1/2 (V vs SCE) |
|---|---|---|---|
| 2a | 309, 383 | 23 (309) | – |
| 2b | 328 | 58 (328) | −0.15 |
| 2c | 293, 499 | 63 (293) | −0.08, −0.14, −0.12 |
aValues in bold represent the highest absorption (λmax) maxima.
The UV–vis absorption spectra were recorded from a CH3CN solution (1.0 × 10−5 M) of the macrocycles at room temperature. The electronic absorption spectra (Figure 6) of the macrocycles 2a–2c exhibit intense bands at λmax (ε) 309 nm (2.3 × 104 M−1 cm−1), 383 nm (6.8 × 103 M−1 cm−1) for 2a; λmax (ε) = 328 nm (5.8 × 104 M−1 cm−1) for 2b and λmax (ε) = 293 nm (6.3 × 104 M−1 cm−1), 499 nm (3.2 × 104 M−1 cm−1) for 2c. The peaks in the ranges of 293–309 nm and 328–499 nm can be ascribed to both intra- and intermolecular π–π* and metal-to-ligand charge-transfer transitions associated with the capped p-cymene–ruthenium moiety, respectively. The electrochemical behavior of the macrocycles (2a–2c) was examined by using a Pt-disk electrode in dichloromethane with 0.1 M n-Bu4NPF6 as the supporting electrolyte and at 100 mV s−1 scan rate. The obtained redox responses of the macrocycles were found to be entirely different. Macrocycle 2c shows three well-anchored quasi-reversible reductions at −0.39, −1.09 and −1.54 V and their corresponding anodic peak current at −0.16, −0.81 and −1.38 V (Figure 6). On the other hand, complex 2b showed a single quasi-reversible redox response of the cathodic peak current at −1.53 V and the anodic peak counterpart potential approximately at −1.24 V (Figure S7, Supporting Information File 1). Surprisingly, macrocycle 2a showed no anodic or cathodic peak current intensity within the range of +0.2 to −0.2 V, which implies the electrochemical inertness of 2a (Figure S7, Supporting Information File 1). The observed anodic and cathodic peak currents of 2c are almost equal, indicating the high chemical stability on the time scale of the voltammetry experiments. Notably, the cathodic peak current intensity of 2b is much higher than its anodic peak current, and this can be attributed to the deposition of macrocycles on the electrode surface. Based on the electrochemical activity of other reported ruthenium complexes, the observed cathodic peaks are roughly assigned to one-electron reductions of the bridged quinonato or diphenyloxamidato ligands, and the oxidation is attributed to the Ru(II)/Ru(III) redox couples [16,17].
![[1860-5397-8-34-6]](/bjoc/content/figures/1860-5397-8-34-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: UV–vis absorption spectrum (left) of macrocycles (2a–2c) recorded in CH3CN at 298 K, and cyclic voltammogram of 2c (right) performed in CH2Cl2/0.1 M (n-Bu)4NPF6 with a scan rate of 100 mV s−1 versus SCE at 298 K.
Figure 6: UV–vis absorption spectrum (left) of macrocycles (2a–2c) recorded in CH3CN at 298 K, and cyclic vol...
Conclusion
In conclusion, we synthesized two new tetranuclear macrocycles 2a, 2b and an octanuclear molecular prism 2c, by coordination-driven self-assembly of an imidazole-based tetratopic donor linker L in combination with Ru(II)-based binuclear “clip” acceptors. All three of the self-assembled macrocycles were fully characterized by multinuclear (1H, 13C and 19F) NMR, IR and ESIMS spectroscopic studies. In addition, the formation of a tetranuclear structure of 2a was unambiguously established by analysis of single-crystal X-ray diffraction data. Though the formation of discrete structures from pyridyl donors has been well established, the quantitative formation of 2a, 2b and 2c from an imidazole-type donor is new and suggests the feasibility of using imidazole derivatives as potential building units. The lengths of 1a and 1b fit well with the distance between two imidazole units in L to form the entropically favored [2 + 1] self-assembled structures 2a and 2b, respectively. As the distance between the two Ru(II) in 1c is greater compared to the distance between the neighboring imidazole units in L, a similar reaction with 1c allowed the formation of the 3D cage 2c. The use of imidazole-based donor linkers in combination with organometallic half-sandwich ruthenium(II) precursors may generate a wide variety of complex molecular architectures with interesting functional properties.
Experimental
Materials and methods
The acceptor clips [Ru2(μ-η4-C2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1a), [Ru2(μ-η4-N,N'-diphenyloxamidato)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1b) and [Ru2(μ-η4-C6H2O4)(MeOH)2(η6-p-cymene)2](O3SCF3)2 (1c) were synthesized under a dry nitrogen atmosphere by means of a standard Schlenk technique following the reported procedures [18-20]. The solvents were dried and distilled according to the standard literature procedures. 1,2,4,5-tetrabromobenzene and α-phellandrene were purchased from commercial sources and used without further purification. 1,2,4,5-Tetrakis(imidazol-1-yl)benzene (L) was synthesized by following the reported procedure [21]. NMR spectra were recorded on a Bruker 400 MHz spectrometer. The chemical shifts (δ) in the 1H NMR spectra are reported in ppm relative to tetramethylsilane (Me4Si) as the internal standard (0.0 ppm) or the proton resonance resulting from incomplete deuteration of the NMR solvents: CD3CN (1.94), CD3OD (3.33) and CD2Cl2 (5.33). 19F NMR were recorded at 376.5 MHz and the chemical shifts (δ) are reported in ppm relative to the external standard Cl3CF (0.00). ESIMS experiments were performed on a Bruker Daltonics (Esquire 300 Plus ESI model) with standard spectroscopic grade solvents CH3CN or CH3OH. IR spectra were recorded on a Bruker ALPHA FT-IR spectrometer. Electronic absorption studies were carried out on a Perkin Elmer LAMBDA 750 UV–vis spectrophotometer. The electrochemical measurements were performed in a three-electrode system consisting of a platinum electrode, a glassy-carbon counter electrode and a standard calomel reference electrode. All the potentials of the macrocycles (1.0 × 10−3 M in DCM) were measured with 0.1 M n-Bu4NPF6 as the supporting electrolyte at room temperature with a scan rate of 100 mV s−1.
General procedure for the synthesis of 2a–2c: To a solid form of the tetratopic donor L was added separately a clear solution of the binuclear acceptor clip (1a–1c) in methanol (4 mL) in 1:2 molar ratio, and the mixture was stirred at room temperature for 24 h in a closed 4 mL glass vial. Immediate consumption of the suspended tetratopic donor (L) and significant visual color changes from light yellow to intense yellow for 2a and 2b or purple to deep red for 2c evidenced the progress of the reactions. The solvent was removed under vacuum and the residue was dissolved in the minimum amount of dichloromethane (DCM) (~2 mL). Diethyl ether was mixed with concentrated clear solutions to precipitate out the expected macrocycles in pure form.
2a: Acceptor clip 1a (29.5 mg, 0.032 mmol) and tetratopic donor L (5.5 mg, 0.016 mmol) were stirred in methanol (4 mL) to obtain 2a. Isolated yield: 85%. 1H NMR (400 MHz, CD2Cl2/CD3OD) δ 8.10 (s, 2H, Ha-phenyl), 7.47 (s, 4H, Hc-imidazole), 7.37 (s, 4H, Hd-imidazole), 6.95 (s, 2H, Hb-imidazole), 5.81 (d, J = 6.0 Hz, 8H, Hα-cymene), 5.70 (d, J = 6.0 Hz, 8H, Hβ-cymene), 2.84–2.77 (septet, 4H, H2-cymene), 2.23 (s, 12H, H3-cymene), 1.33 (d, 24H, H1-cymene); 19F NMR (376.5 MHz, CD2Cl2/CD3OD) δ −81.03 ppm; 13C NMR (100 MHz, CD3COCD3) δ 171.65, 140.12, 136.08, 131.62, 129.14, 126.36, 124.65, 101.55, 98.10, 82.85, 81.12, 54.51, 31.56, 22.03, 17.79; IR (neat) ν (cm−1): 3128.8 (w, C=C of aromatic), 1624.6 (s, CO-oxalate), 1254.9 (s, C–F of CF3); ESIMS (m/z): 1907.16 [2a − O3SCF3−]+, 879.0 [2a − 2O3SCF3−]2+; UV–vis (1.0 × 10−5 M, CH3CN) λmax nm (ε): 309 (2.3 × 104 M−1 cm−1), 383 (6.8 × 103 M−1 cm−1); Anal. calcd for C66H70F12N8O20Ru4S4: C, 38.56; H, 3.43; N, 5.45; found: C, 38.84; H, 3.68; N, 5.71.
2b: Acceptor clip 1b (34.3 mg, 0.032 mmol) and tetratopic donor L (5.5 mg, 0.016 mmol) were stirred in methanol (4 mL) to obtain 2b. Isolated yield: 88%. 1H NMR (400 MHz, CD3OD) δ 8.19 (s, 4H, Hc-imidazole), 8.17 (s, 4H, Hd-imidazole), 7.82 (s, 4H, Hb-imidazole), 7.46–7.21 (m, 20H, N-phenyl), 7.17 (s, 2H, Ha-imidazole), 5.63–5.14 (m, 16H, Hα,β-cymene), 2.50–2.48 (septet, 4H, H2-cymene), 1.71 (s, 12H, H3-cymene), 1.12 (d, 24H, H1-cymene); 19F NMR (376.5 MHz, CD3OD) δ −79.84; 13C NMR (100 MHz, CD3NO2) δ 170.89, 147.02, 139.76, 135.54, 132.74, 129.87, 128.63, 127.03, 125.45, 123.82, 123.00, 119.81, 104.32, 99.43, 85.27, 83.99, 82.23, 81.35, 65.70, 31.47, 22.30, 20.99, 17.16, 14.70; IR (neat) ν (cm−1): 3136 (w, C=C of aromatic), 1602 (s, CO-oxamide), 1262 (s, C–F of CF3); ESIMS (m/z): 1029.53 [2b − 2O3SCF3−]2+, 637.27 [2b − 3O3SCF3−]3+; UV–vis (1.0 × 10−5 M, CH3CN) λmax nm (ε): 328 (5.8 × 104 M−1 cm−1); Anal. calcd for C90H90F12N12O16Ru4S4: C, 45.88; H, 3.85; N, 7.13; found: C, 46.59; H, 4.17; N, 7.43;
2c: Acceptor clip 1c (31.1 mg, 0.032 mmol) and tetratopic donor L (5.5 mg, 0.016 mmol) were stirred in methanol (4 mL) to obtain 2c. Isolated yield: 80%.1H NMR (400 MHz, CD3CN) δ 7.89 (s, 4H, Ha-phenyl), 7.40 (s, 8H, Hc-imidazole), 7.36 (s, 8H, Hd-imidazole), 6.96 (s, 8H, Hb-imidazole), 5.94 (d, J = 5.6 Hz, 16H, Hα-cymene), 5.88 (s, 4H, H4-quinone), 5.75 (d, J = 6.0 Hz, 16H, Hβ-cymene), 2.86–2.79 (septet, 8H, H2-cymene), 2.22 (s, 24H, H3-cymene), 1.31 (d, 48H, H1-cymene); 19F NMR (376.5 MHz, CD3CN) δ −79.08; 13C NMR (100 MHz, CD3NO2) δ 183.92, 140.62, 134.75, 131.45, 124.09, 122.92, 119.74, 103.76, 99.10, 84.88, 83.65, 79.47, 78.00, 31.78, 22.27, 17.70; IR (neat) ν (cm−1): 3128.8 (w, C=C of aromatic), 1521.2 (s, CO-oxalate), 1252.1 (s, C–F of CF3); ESIMS (m/z): 2008.0 [2c − 2O3SCF3−]2+, 1288.0 [2c − 3O3SCF3−]3+, 928.9 [2c − 4O3SCF3−]4+; UV–vis (1.0 × 10−5 M, CH3CN) λmax nm (ε): 293 (6.3 × 104 M−1 cm−1), 499 (3.2 × 104 M−1 cm−1); Anal. calcd for C148H148F24N16O40Ru8S8: C, 41.23; H, 3.46; N, 5.20; found: C, 41.83; H, 4.16; N, 5.60.
X-ray data collection and structure refinements: Crystallographic data for 2a were collected on a Bruker SMART APEX CCD diffractometer with the SMART/SAINT software [22]. X-ray-quality crystals were mounted on a glass fiber with traces of viscous oil. Intensity data were collected by using graphite-monochromatized Mo Kα radiation (0.7107 Å) at 150 K. The structures were solved by direct methods using SHELX-97 incorporated in WinGX [23-27]. Empirical absorption corrections were applied with SADABS [25]. All nonhydrogen atoms were refined with anisotropic displacement coefficients. Hydrogen atoms were assigned isotropic displacement coefficients, U(H) = 1.2U(C) or 1.5U (C-methyl), and their coordinates were allowed to ride on their respective carbons.
CCDC-845980 (for complex 2a) contains the supplementary crystallographic data reported in this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Center via http://www.ccdc.cam.ac.uk/data_request/cif.
Crystallographic data: C66H70F12N8O20Ru4S4, Mr = 2055.86, tetragonal, space group I41/a, a = 27.975(5) Å, b = 27.975(5) Å, c = 48.786(17) Å, α = 90°, β = 90°, γ = 90°, V = 38179(16) Å3, Z = 16, ρcalcd. = 1.431 g cm−1, Mo Kα radiation (graphite monochromatic, λ = 0.71073 Å), T = 150 K, final R indices [I > 2σ(I)]: R1 = 0.1152 (5366), wR2 = 0.4337 (18769).
Acknowledgements
S.S. and D.S. gratefully acknowledge the Council of Scientific and Industrial Research, New Delhi, India for the award of a research fellowship. P.S.M. thanks the Department of Science and Technology (DST), India, for financial support. S.S. sincerely thanks Mr. R. Sathish Kumar (SSCU, IISc) and Mr. Yogesh P. Patil (IPC, IISc) for their kind help in the X-ray data collection and structure solving. S.S. thanks Mr. Tridip (IPC, IISc) for his assistance in the cyclic voltammetry study. The authors are grateful to Johnson Matthey Pvt. Ltd. U.K. for their generous donation of RuCl3.
References
-
Northrop, B. H.; Yang, H.-B.; Stang, P. J. Chem. Commun. 2008, 5896–5908. doi:10.1039/b811712h
Return to citation in text: [1] -
Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340
Return to citation in text: [1] -
Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. Chem. Rev. 2011, 111, 6810–6918. doi:10.1021/cr200077m
Return to citation in text: [1] -
Shanmugaraju, S.; Bar, A. K.; Chi, K.-W.; Mukherjee, P. S. Organometallics 2010, 29, 2971–2980. doi:10.1021/om100202c
Return to citation in text: [1] -
Shanmugaraju, S.; Joshi, S. A.; Mukherjee, P. S. Inorg. Chem. 2011, 50, 11736–11745. doi:10.1021/ic201745y
Return to citation in text: [1] -
Therrien, B. Eur. J. Inorg. Chem. 2009, 2445–2453. doi:10.1002/ejic.200900180
Return to citation in text: [1] -
Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j
Return to citation in text: [1] -
Wang, M.; Vajpayee, V.; Shanmugaraju, S.; Zheng, Y.-R.; Zhao, Z.; Kim, H.; Mukherjee, P. S.; Chi, K.-W.; Stang, P. J. Inorg. Chem. 2011, 50, 1506–1512. doi:10.1021/ic1020719
Return to citation in text: [1] -
Shanmugaraju, S.; Bar, A. K.; Mukherjee, P. S. Inorg. Chem. 2010, 49, 10235–10237. doi:10.1021/ic101823s
Return to citation in text: [1] -
Shanmugaraju, S.; Bar, A. K.; Joshi, S. A.; Patil, Y. P.; Mukherjee, P. S. Organometallics 2011, 30, 1951–1960. doi:10.1021/om2000019
Return to citation in text: [1] -
Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062
Return to citation in text: [1] -
Fujita, M.; Yu, S.-Y.; Kusukawa, T.; Funaki, H.; Ogura, K.; Yamaguchi, K. Angew. Chem., Int. Ed. 1998, 37, 2082–2085. doi:10.1002/(SICI)1521-3773(19980817)37:15<2082::AID-ANIE2082>3.0.CO;2-0
Return to citation in text: [1] -
ArgusLab 4.0; Planaria Software LLC: Seattle, WA, http://www.arguslab.com.
Return to citation in text: [1] -
Govindaswamy, P.; Linder, D.; Lacour, J.; Süss-Fink, G.; Therrien, B. Chem. Commun. 2006, 4691–4693. doi:10.1039/b610155k
Return to citation in text: [1] -
Amijs, C. H. M.; van Klink, G. P. M.; van Koten, G. Dalton Trans. 2006, 308–327. doi:10.1039/b505354d
Return to citation in text: [1] -
Mattsson, J.; Govindaswamy, P.; Renfrew, A. K.; Dyson, P. J.; Stěpnička, P.; Süss-Fink, G.; Therrien, B. Organometallics 2009, 28, 4350–4357. doi:10.1021/om900359j
Return to citation in text: [1] -
Kitagawa, S.; Kawata, S. Coord. Chem. Rev. 2002, 224, 11–34. doi:10.1016/S0010-8545(01)00369-1
Return to citation in text: [1] -
Barry, N. P. E.; Govindaswamy, P.; Furrer, J.; Süss-Fink, G.; Therrien, B. Inorg. Chem. Commun. 2008, 11, 1300–1303. doi:10.1016/j.inoche.2008.08.007
Return to citation in text: [1] -
Zhang, W.-Z.; Han, Y.-F.; Lin, Y.-J.; Jin, G.-X. Dalton Trans. 2009, 8426–8431. doi:10.1039/b909357e
Return to citation in text: [1] -
Yan, H.; Süss-Fink, G.; Neels, A.; Stoeckli-Evans, H. J. Chem. Soc., Dalton Trans. 1997, 4345–4350. doi:10.1039/A704658H
Return to citation in text: [1] -
Rit, A.; Pape, T.; Hepp, A.; Hahn, F. E. Organometallics 2011, 30, 334–347. doi:10.1021/om101102j
Return to citation in text: [1] -
SMART/SAINT; Bruker AXS, Inc.: Madison, WI, 2004.
Return to citation in text: [1] -
Sheldrick, G. M. SHELX-97, Program for the Solution and Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1998.
Return to citation in text: [1] -
WinGX: An Integrated System of Windows Programs for the Solution, Refinement; Analysis for Single Crystal X-ray Diffraction Data, Version 1.65.04; Department of Chemistry: University of Glasgow: Glasgow, UK, 2003.
Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020
Return to citation in text: [1] -
Sheldrick, G. M. SADABS, Bruker Nonius Area Detector Scaling and Absorption Correction; Version 2.05; University of Göttingen: Göttingen, Germany, 1999.
Return to citation in text: [1] [2] -
ORTEP-3 for Windows, Version, 1.08.
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117
Return to citation in text: [1] -
Spek, A. L. Acta Crystallogr. 1990, A46, C34.
Return to citation in text: [1]
| 1. | Northrop, B. H.; Yang, H.-B.; Stang, P. J. Chem. Commun. 2008, 5896–5908. doi:10.1039/b811712h |
| 2. | Yoshizawa, M.; Klosterman, J. K.; Fujita, M. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. doi:10.1002/anie.200805340 |
| 12. | Fujita, M.; Yu, S.-Y.; Kusukawa, T.; Funaki, H.; Ogura, K.; Yamaguchi, K. Angew. Chem., Int. Ed. 1998, 37, 2082–2085. doi:10.1002/(SICI)1521-3773(19980817)37:15<2082::AID-ANIE2082>3.0.CO;2-0 |
| 11. | Northrop, B. H.; Chercka, D.; Stang, P. J. Tetrahedron 2008, 64, 11495–11503. doi:10.1016/j.tet.2008.08.062 |
| 6. | Therrien, B. Eur. J. Inorg. Chem. 2009, 2445–2453. doi:10.1002/ejic.200900180 |
| 7. | Han, Y.-F.; Jia, W.-G.; Yu, W.-B.; Jin, G.-X. Chem. Soc. Rev. 2009, 38, 3419–3434. doi:10.1039/b901649j |
| 8. | Wang, M.; Vajpayee, V.; Shanmugaraju, S.; Zheng, Y.-R.; Zhao, Z.; Kim, H.; Mukherjee, P. S.; Chi, K.-W.; Stang, P. J. Inorg. Chem. 2011, 50, 1506–1512. doi:10.1021/ic1020719 |
| 9. | Shanmugaraju, S.; Bar, A. K.; Mukherjee, P. S. Inorg. Chem. 2010, 49, 10235–10237. doi:10.1021/ic101823s |
| 10. | Shanmugaraju, S.; Bar, A. K.; Joshi, S. A.; Patil, Y. P.; Mukherjee, P. S. Organometallics 2011, 30, 1951–1960. doi:10.1021/om2000019 |
| 23. | Sheldrick, G. M. SHELX-97, Program for the Solution and Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1998. |
| 24. |
WinGX: An Integrated System of Windows Programs for the Solution, Refinement; Analysis for Single Crystal X-ray Diffraction Data, Version 1.65.04; Department of Chemistry: University of Glasgow: Glasgow, UK, 2003.
Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020 |
| 25. | Sheldrick, G. M. SADABS, Bruker Nonius Area Detector Scaling and Absorption Correction; Version 2.05; University of Göttingen: Göttingen, Germany, 1999. |
| 26. |
ORTEP-3 for Windows, Version, 1.08.
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 27. | Spek, A. L. Acta Crystallogr. 1990, A46, C34. |
| 3. | Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. Chem. Rev. 2011, 111, 6810–6918. doi:10.1021/cr200077m |
| 4. | Shanmugaraju, S.; Bar, A. K.; Chi, K.-W.; Mukherjee, P. S. Organometallics 2010, 29, 2971–2980. doi:10.1021/om100202c |
| 5. | Shanmugaraju, S.; Joshi, S. A.; Mukherjee, P. S. Inorg. Chem. 2011, 50, 11736–11745. doi:10.1021/ic201745y |
| 25. | Sheldrick, G. M. SADABS, Bruker Nonius Area Detector Scaling and Absorption Correction; Version 2.05; University of Göttingen: Göttingen, Germany, 1999. |
| 16. | Mattsson, J.; Govindaswamy, P.; Renfrew, A. K.; Dyson, P. J.; Stěpnička, P.; Süss-Fink, G.; Therrien, B. Organometallics 2009, 28, 4350–4357. doi:10.1021/om900359j |
| 17. | Kitagawa, S.; Kawata, S. Coord. Chem. Rev. 2002, 224, 11–34. doi:10.1016/S0010-8545(01)00369-1 |
| 21. | Rit, A.; Pape, T.; Hepp, A.; Hahn, F. E. Organometallics 2011, 30, 334–347. doi:10.1021/om101102j |
| 15. | Amijs, C. H. M.; van Klink, G. P. M.; van Koten, G. Dalton Trans. 2006, 308–327. doi:10.1039/b505354d |
| 14. | Govindaswamy, P.; Linder, D.; Lacour, J.; Süss-Fink, G.; Therrien, B. Chem. Commun. 2006, 4691–4693. doi:10.1039/b610155k |
| 18. | Barry, N. P. E.; Govindaswamy, P.; Furrer, J.; Süss-Fink, G.; Therrien, B. Inorg. Chem. Commun. 2008, 11, 1300–1303. doi:10.1016/j.inoche.2008.08.007 |
| 19. | Zhang, W.-Z.; Han, Y.-F.; Lin, Y.-J.; Jin, G.-X. Dalton Trans. 2009, 8426–8431. doi:10.1039/b909357e |
| 20. | Yan, H.; Süss-Fink, G.; Neels, A.; Stoeckli-Evans, H. J. Chem. Soc., Dalton Trans. 1997, 4345–4350. doi:10.1039/A704658H |
© 2012 Shanmugaraju et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)