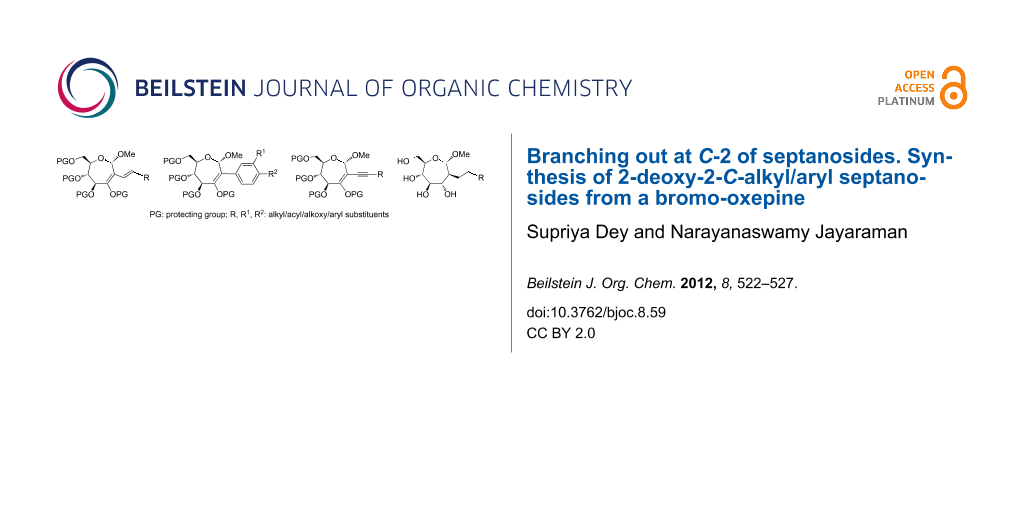
Abstract
This paper deals with the synthesis of 2-deoxy-2-C-alkyl/aryl septanosides. A range of such septanoside derivatives was synthesized by using a common bromo-oxepine intermediate, involving C–C bond forming organometallic reactions. Unsaturated, seven-membered septanoside vinyl bromides or bromo-oxepines, obtained through a ring expansion methodology of the cyclopropane derivatives of oxyglycals, displayed a good reactivity towards several acceptor moieties in C–C bond forming Heck, Suzuki and Sonogashira coupling reactions, thus affording 2-deoxy-2-C-alkyl/aryl septanosides. Whereas Heck and Sonogashira coupling reactions afforded 2-deoxy-2-C-alkenyl and -alkynyl derivatives, respectively, the Suzuki reaction afforded 2-deoxy-2-C-aryl septanosides. Deprotection and reduction of the 2-deoxy-2-alkenyl derivative afforded the corresponding 2-deoxy-2-C-alkyl septanoside free of protecting groups. The present study illustrates the reactivity of bromo-oxepine in the synthesis of hitherto unknown septanosides, branching out at C-2, through C–C bond formation with alkyl and aryl substituents.

Graphical Abstract
Introduction
Septanoses and septanosides are unnatural, seven-membered cyclic sugars [1]. Methods of preparation and the exploration of the properties of these unnatural sugars are of high interest [2]. An early isolation of septanose was achieved through cyclization of generic hexose sugars, which afforded minor amounts of septanose, along with furanose and pyranose, which formed in major amounts [3]. Synthetic approaches to septanoses have been explored in many instances, for example, (i) hemiacetal or acetal formation from a linear precursor containing aldehyde and an appropriately positioned hydroxyl group [4-8]; (ii) Knoevenagel-type condensation of sugar aldehyde with active methylene compounds [9,10]; (iii) ring-closing metathesis reactions of appropriately installed diene derivatives [11-13]; (iv) ring expansion of 1,2-cyclopropanated sugars [14-17]; (v) Baeyer–Villiger oxidation of inositol derivatives [18,19] and (vi) electrophile-induced cyclization [20]. We recently developed a new methodology to prepare septanosides, which involved a sequence of dihalocarbene insertion on to an oxyglycal, ring opening of the cyclopropyl moiety with a nucleophile, and oxidation and reduction reactions, so as to permit the expansion of six-membered pyranoses to seven-membered septanosides [21-23]. Features of this methodology include the formation of vinyl halide, vinyl ether, diketone and diol intermediates, which are potential sites for varied types of functionalizations. While exploring such features, we undertook the preparation of septanoside derivatives that are branched out at C-2, so as to afford 2-deoxy-2-C-alkyl/aryl derivatives, through C–C bond formations mediated by organometallic reagents. Details of the preparation of 2-deoxy-2-C-aryl/alkyl septanosides are described herein.
Results and Discussion
The methodology of septanoside preparation starting from an oxyglycal is shown in Figure 1 [21]. The oxygen at C-2 of oxyglycal I was retained throughout until the septanoside V was obtained. More importantly, vinyl halide III and diketone IV also form as intermediates of the reaction and these intermediates provide an avenue to expand the scope of the reaction sequence.
![[1860-5397-8-59-1]](/bjoc/content/figures/1860-5397-8-59-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthetic route to transform oxyglycal I to a septanoside V.
Figure 1: Synthetic route to transform oxyglycal I to a septanoside V.
In the present work, we envisaged that III would form as a synthon to implement C–C bond forming reactions. Vinyl halide 2 was synthesized through a ring-expansion reaction of cyclopropanated adduct 1 (Scheme 1), as reported previously [21]. The reactivity at C-2 of 2 was examined by the chosen organometallic reactions, namely, Heck, Suzuki and Sonogashira coupling reactions. Heck coupling reactions [24,25] were undertaken first. Thus, the reaction of bromo-oxepine 2 with methyl acrylate was performed, in the presence of Pd(OAc)2 (10 mol %) and Cs2CO3 in 1,4-dioxane, at 98 °C (Scheme 1), to afford diene 3, in 70% yield. The presence of doublets at 7.80 and 5.97 ppm (J = 16.0 Hz) in the 1H NMR spectrum and signals at 136.3 ppm and 119.5 ppm in the 13C NMR spectrum confirmed the formation of 3.
![[1860-5397-8-59-i1]](/bjoc/content/inline/1860-5397-8-59-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction conditions: (i) NaOMe, PhMe, reflux, 8 h; (ii) methyl acrylate (for 3); tert-butyl acrylate (for 4); H2C=CHCOO(CH2)5OCOCH=CH2 (for 5; 2/substrate = 1:0.57); styrene (for 6); α-methyl styrene (for 7); Pd(OAc)2 (10 mol %), Cs2CO3 (1.5 molar equiv), 1,4-dioxane, 98 °C, 72 h.
Scheme 1: Reaction conditions: (i) NaOMe, PhMe, reflux, 8 h; (ii) methyl acrylate (for 3); tert-butyl acrylat...
Having realized the synthesis of one product, reactions of 2 were performed with a few other substrates, namely, tert-butyl acrylate, a substrate presenting two acrylates within the molecule, styrene, and α-methyl styrene (Scheme 1). Reactions with these substrates also afforded the diene products 4–7, in good yields. The anticipated two Heck coupling reactions with the substrate that presents two acrylates, could not be achieved, rather only the mono-Heck coupling product 5 was obtained. Alternative reaction conditions were attempted, for example, by using Pd(PPh3)2Cl2 (10 mol %), instead of Pd(OAc)2, while keeping other parameters of the reaction uniform, yet the double-Heck coupling product was not observed. The newly generated exocyclic olefin protons resonated as two distinct doublets in 4 at 7.72 and 5.88 ppm (J = 16.4 Hz); in 5 at 7.79 and 5.93 ppm (J = 16.4 Hz) and in 6 at 7.19 and 6.66 ppm (J = 16.8 Hz). Further, the exocyclic double-bond carbon nuclei resonated at ~136–130 and ~122 ppm in the 13C NMR spectra of 4–6. The reactions afforded only the (E)-isomer. Interestingly, when the reaction was performed with α-methyl styrene, product 7, with an exocyclic double bond isomerization to a terminal double bond was observed. The appearance of two singlets at 5.30 and 5.14 ppm in the 1H NMR spectrum indicated the presence of two vinylic protons in 7. On the other hand, the exocyclic methylene moiety at C-2 in 7 appeared as two distinct doublets (3.88, 3.08 ppm, J = 14.4 Hz) in the 1H NMR spectrum. Further structural assignments of 7 were performed through COSY and HSQC experiments.
Following the Heck coupling reactions on bromo-oxepine 2, efforts were undertaken to implement C–C bond forming Suzuki and Sonogashira coupling reactions. Suzuki reactions were undertaken by involving phenylboronic acid and substituted phenylboronic acids [26,27], in the presence of Pd(OAc)2 (10 mol %) and Cs2CO3 in 1,4-dioxane at 98 °C (Scheme 2). The reactions afforded septanosides 8–10, which are derivatized with a phenyl substituent at C-2, in moderate yields. The formation of a C–C bond at C-2 in 8–10 was inferred by the observation of shifts of the C-2 nuclei signal at ~129 ppm, which in the case of bromo-oxepine was observed at 114.3 ppm. Analyses of 1H and 13C NMR spectra and mass spectra confirmed the constitution of 8–10.
![[1860-5397-8-59-i2]](/bjoc/content/inline/1860-5397-8-59-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction conditions: (i) phenylboronic acid (for 8); 4-methoxyphenylboronic acid (for 9); 3-methylphenylboronic acid (for 10); Pd(OAc)2 (10 mol %), Cs2CO3 (1.5 molar equiv), 1,4-dioxane, 98 °C, 72 h.
Scheme 2: Reaction conditions: (i) phenylboronic acid (for 8); 4-methoxyphenylboronic acid (for 9); 3-methylp...
The reactivity of bromo-oxepine, the key intermediate of the septanoside synthesis earlier, was explored further in the context of C–C bond formation at C-2, through another versatile C–C bond forming reaction, namely, Sonogashira coupling [28,29]. Reactions of 2 with acetylenes were performed in the presence of Pd(PPh3)2Cl2 (20 mol %) and CuI (10 mol %) in a DMF/THF/Et3N 5:3:2 solvent mixture as the optimized protocol. The use of Pd(OAc)2 as a catalyst or Et3N as the base did not promote the reaction, leading only to the recovery of the starting material. Thus, the reaction of 2 with phenylacetylene and oct-1-yne led to the formation of the corresponding 2-deoxy-2-C-alkynyl septanosides 11 and 12 (Scheme 3) in moderate yields. Prolonging the reaction time and increasing the catalyst loading did not increase the yields, although dehalogenation of 2 to oxepine was found to occur to a minor extent when the reaction time was increased to several days. 13C NMR spectra of 11 and 12 showed resonances for the newly formed C–C bond at 11: 108.4 ppm (C-2) and 95.8 ppm (C≡C-Ph); 12: 109.9 ppm (C-2) and 97.4 ppm (C≡C-C6H13). Further, 1H and 13C NMR spectroscopic and mass spectrometric analyses confirmed the constitutions of 11 and 12.
![[1860-5397-8-59-i3]](/bjoc/content/inline/1860-5397-8-59-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction conditions: (i) phenylacetylene (for 11); oct-1-yne (for 12); Pd(PPh3)2Cl2 (20 mol %), CuI (10 mol %), DMF/THF/Et3N 3:2:1, 98 °C, 72 h.
Scheme 3: Reaction conditions: (i) phenylacetylene (for 11); oct-1-yne (for 12); Pd(PPh3)2Cl2 (20 mol %), CuI...
Having observed a good reactivity of bromo-oxepine 2 in C–C bond forming reactions, we used one of the 2-deoxy-2-C-alkyl derivatives, namely, product 4 for further reactions, leading to a 2-deoxy-2-C-alkyl septanoside containing free hydroxyl groups. Towards this effort, 4 was subjected first to a hydrogenolysis (Pd/C, H2), which afforded D-manno-sept-3-uloside 13 as single diastereomer in good yield (Scheme 4).
![[1860-5397-8-59-i4]](/bjoc/content/inline/1860-5397-8-59-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction conditions: (i) Pd/C (10 %), H2, MeOH, rt, 24 h; (ii) NaBH4, MeOH, 0 °C to rt, 3 h.
Scheme 4: Reaction conditions: (i) Pd/C (10 %), H2, MeOH, rt, 24 h; (ii) NaBH4, MeOH, 0 °C to rt, 3 h.
The configuration of C-2 in 13 was confirmed through HMQC and COSY experiments. A doublet at 4.37 ppm with JH1,H2 of 8.0 Hz indicated a trans-configuration of H-2 with respect to H-1 in 13. The presence of sets of protons in the 1H NMR spectrum, one at 2.05 and 1.78 ppm (multiplet) and the other at 2.21 ppm (t, J = 7.4 Hz), corresponding to exocyclic methylene moieties in 13, resulting from the concomitant reduction of the exocyclic double bond in 4, was also observed. The presence of the ketone functionality in 13 was inferred from the resonance at 208.4 ppm in the 13C NMR spectrum. Subsequent to hydrogenolysis, the treatment of 13 with NaBH4 facilitated the reduction of the keto-moiety to the corresponding alcohol 14, in an excellent yield. The trans-bisequatorial configuration of the hydroxyl groups at C-3 and C-4 in 14 was inferred from a 3JH3,H4 of 12.4 Hz, in the 1H NMR spectrum. On the other hand, the proton at C-2 merged with the exocyclic methylene group, leading to an inability to assess the H-2,H-3 coupling constant in 14. Having defined the configuration of the substituent at C-2 in 13, we infer a trans-configuration of the substituent at C-2 and C-3. The results of mass spectrometric analysis concurred with the constitutions of 13 and 14.
Conclusion
The present study illustrates the effective application of synthetically useful bromo-oxepine for the preparation of hitherto unknown 2-deoxy-2-C-alkyl/aryl septanoside derivatives. C–C bond forming Heck, Suzuki and Sonogashira coupling reactions, with appropriate acrylates, arylboronic acids and alkynes, afforded the respective cross-coupled products in good yields. It is pertinent to note that the implementation of such reactions is known in seven-membered 1,2-oxazepines, so as to secure the corresponding cross-coupling products [30]. Furthermore, one of the 2-deoxy-2-C-alkyl septanoside derivatives was converted to a hydroxyl-group-free methyl 2-deoxy-2-C-alkyl septanoside. The present study illustrates the scope of seven-membered bromo-oxepines as useful substrates for the generation of 2-deoxy-2-C-alkyl/aryl septanosides, in addition to our previous efforts to progress such intermediates to a number septanosides and septanoside-containing di- and tri-saccharides.
Experimental
General
Chemicals were purchased from commercial sources and were used without further purification. Solvents were dried and distilled according literature procedures. Analytical TLC was performed on commercial Merck plates coated with silica gel GF254 (0.25 mm). Silica gel (230–400 mesh) was used for column chromatography. Optical rotations were recorded on a JASCO Model P-1020 polarimeter at the sodium D line at 24 °C. High-resolution mass spectra were obtained from a Q-TOF instrument by the electrospray ionization (ESI) technique. 1H and 13C NMR spectral analyses were performed on 400 MHz and 100 MHz spectrometers, respectively, with the residual solvent signal acting as the internal standard. COSY and HSQC analyses were performed on a 400 MHz NMR spectrometer.
Methyl 2-deoxy-2-C-(2-(tert-butoxycarbonyl)vinyl)-3,4,5,7-tetra-O-benzyl-α-D-arabino-hept-2-enoseptanoside (4): A solution of 2 [21] (0.05 g, 0.07 mmol) in 1,4-dioxane (1 mL) was admixed with Pd(OAc)2 (1 mg, 10 mol %) under a N2 atmosphere, and this was followed by the addition of Cs2CO3 (0.03 g, 0.11 mmol) and tert-butyl acrylate (0.02 mL, 0.153 mmol), in a sealed tube. The reaction mixture was stirred at 98 °C for 72 h, cooled, filtered, diluted with EtOAc (20 mL), washed with water (2 × 30 mL) and brine (2 × 10 mL), dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified (hexane/EtOAc 9:1) to afford 4 (0.038 g, 72%), as an oil. Rf 0.48 (hexane/EtOAc 9:1); [α]D −130.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 16.4 Hz, 1H, -CH=CHCO2t-Bu), 7.33–7.24 (m, 18H, aromatic), 7.10–7.08 (m, 2H, aromatic), 5.88 (d, J = 16.4 Hz, 1H, -CH=CHCO2t-Bu), 5.36 (s, 1H, H-1), 4.69–4.56 (m, 4H, PhCH2), 4.48 (d, J = 12.0 Hz, 1H, PhCH2), 4.43 (d, J = 12.0 Hz, 1H, PhCH2), 4.33 (d, J = 12.0 Hz, 1H, PhCH2), 4.24 (d, J = 12.0 Hz, 1H, PhCH2), 4.21–4.17 (m, 2H, H-4 and H-6), 3.75 (dd, J = 8.4, 1.4 Hz, 1H, H-5), 3.63–3.57 (br, 1H, H-7a), 3.53–3.52 (br, 1H, H-7b), 3.51 (s, 3H, OMe), 1.47 (s, 9H, t-Bu); 13C NMR (100 MHz, CDCl3) δ 166.6 (C=O), 158.7 (C-3), 138.2–137.0 (aromatic), 136.4 (-CH=CHCO2t-Bu), 128.4–127.5 (aromatic ), 124.4 (C-2), 121.9 (-CH=CHCO2t-Bu), 100.0 (C-1), 80.1 (C-5), 79.9 (C-4), 73.0 (PhCH2), 72.8 (PhCH2), 72.0 (PhCH2), 71.2 (PhCH2), 71.0 (C-6), 70.8 (C-7), 55.5 (OMe), 28.1 (t-Bu); HRMS–ESI (m/z): [M + Na]+ calcd for 715.3247; found, 715.3245.
Methyl 2-deoxy-2-C-(2-phenylallyl)-3,4,5,7-tetra-O-benzyl-α-D-arabino-hept-2-enoseptanoside (7): A solution of 2 [21] (0.05 g, 0.07 mmol) in 1,4-dioxane (1 mL) was admixed with Pd(OAc)2 (1 mg, 10 mol %) under a N2 atmosphere, and was followed by the addition of Cs2CO3 (0.03 g, 0.11 mmol) and α-methyl styrene (0.01 mL, 0.09 mmol) in a sealed tube. The reaction mixture was stirred at 98 °C for 72 h, cooled, filtered, diluted with EtOAc (20 mL), washed with water (2 × 30 mL) and brine (2 × 10 mL), dried (Na2SO4) and concentrated in vacuo. The crude reaction mixture was purified (hexane/EtOAc 92:8) to afford 7 (0.042 g, 80%), as an oil. Rf 0.60 (hexane/EtOAc 9:1); [α]D −58.8 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.6 Hz, 2H, aromatic), 7.33–7.21 (m, 21H, aromatic), 7.18 (d, J = 4.8 Hz, 2H, aromatic), 5.30 (app. s, 1H, CHH=CPh), 5.14 (app. s, 1H, CHH=CPh), 4.98 (s, 1H, H-1), 4.58 (d, J = 12.4 Hz, 2H, PhCH2), 4.44 (d, J = 12.0 Hz, 2H, PhCH2), 4.31 (d, J = 10.8 Hz, 2H, PhCH2), 4.22 (d, J = 11.6 Hz, 1H, PhCH2), 4.08–4.03 (band, 3H, H-4, H-6 and PhCH2), 3.88 (d, J = 14.4 Hz, 1H, -CHH C(Ph)=CH2), 3.61 (dd, J = 9.2, 1.6 Hz, 1H, H-5), 3.55 (dd, J = 10.4, 6.4 Hz, 1H, H-7a), 3.49 (dd, J = 8.8, 2.0 Hz, 1H, H-7b), 3.36 (s, 3H, OMe), 3.08 (d, J = 14.4 Hz, 1H, -CHH C(Ph)=CH2); 13C NMR (100 MHz, CDCl3) δ 150.9 (C-3), 146.3 (CH2-C(Ph)=CH2), 141.0–137.3 (aromatic), 128.3–126.6 (aromatic), 126.5 (C-2), 114.1 (C-10), 101.0 (C-1), 80.8 (C-5), 76.3 (C-4), 72.9 (PhCH2), 72.0 (PhCH2), 71.7 (PhCH2), 71.3 (PhCH2), 71.2 (C-7), 70.0 (C-6), 55.7 (OMe) 33.2 (-CH2-C(Ph)=CH2); HRMS–ESI (m/z): [M + Na]+ calcd for 705.3192; found, 705.3193.
Methyl 2-deoxy-2-C-(p-methoxyphenyl)-3,4,5,7-tetra-O-benzyl-α-D-arabino-hept-2-enoseptanoside (9): A solution of 2 [21] (0.05 g, 0.07 mmol) in 1,4-dioxane (1 mL) was admixed with Pd(OAc)2 (1 mg, 10 mol %) under a N2 atmosphere, and was followed by the addition of Cs2CO3 (0.03 g, 0.11 mmol) and 4-methoxyphenylboronic acid (0.012 g, 0.07 mmol), in a sealed tube. The reaction mixture was stirred at 98 °C for 72 h, cooled, filtered, diluted with EtOAc (20 mL), washed with water (2 × 30 mL) and brine (2 × 10 mL), dried (Na2SO4) and concentrated in vacuo. The crude reaction mixture was purified (hexane/EtOAc 8:2) to afford 9 (0.033 g, 64%), as an oil. Rf 0.60 (hexane/EtOAc 8:2); [α]D −9.8 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.38–7.11 (m, 20H, aromatic), 6.88 (d, J = 7.6 Hz, 2H, aromatic), 6.83 (d, J = 8.8 Hz, 2H, aromatic), 5.37 (s, 1H, H-1), 4.81 (d, J = 12.4 Hz, 1H, PhCH2), 4.64–4.49 (m, 3H, PhCH2), 4.40 (d, J = 11.6 Hz, 2H, PhCH2), 4.32–4.29 (br, 1H, H-6), 4.28 (app. d, J = 11.2 Hz, 1H, H-4), 4.23 (s, 2H, PhCH2), 3.80 (s, 3H, OMe), 3.77–3.74 (br, 1H, H-5), 3.66 (dd, J = 10.6, 6.4 Hz, 1H, H-7a), 3.58 (dd, J = 10.6, 2.4 Hz, 1H, H-7b), 3.33 (s, 3H, OMe); 13C NMR (100 MHz, CDCl3) δ 158.4 (aromatic), 152.3 (C-3), 138.4–137.3 (aromatic), 130.9 (aromatic), 129.0 (C-2), 128.4–127.4 (aromatic), 113.1 (aromatic), 102.2 (C-1), 80.7 (C-5), 78.3 (C-4), 73.0 (PhCH2), 72.6 (PhCH2), 72.0 (PhCH2), 71.2 (C-6), 71.1 (C-7), 55.9 (OMe), 55.2 (-C6H4OMe); HRMS–ESI (m/z): [M + Na]+ calcd for 695.2985; found, 695.2983.
Methyl 2-deoxy-2-C-(octyn-1-yl)-3,4,5,7-tetra-O-benzyl-α-D-arabino-hept-2-enoseptanoside (12): A solution of 2 [21] (0.05 g, 0.07 mmol) in DMF/THF/Et3N 5:3:2 (1 mL) was admixed with Pd(PPh3)2Cl2 (0.01 g, 20 mol %) under a N2 atmosphere, and was followed by the addition of CuI (0.012 g, 10 mol %) and 1-octyne (0.023 mL, 0.14 mmol), in a sealed tube. The reaction mixture was stirred at 98 °C for 72 h, cooled, filtered, diluted with EtOAc (20 mL), washed with water (2 × 30 mL) and brine (2 × 10 mL), dried (Na2SO4) and concentrated in vacuo. The crude reaction mixture was purified (hexane/EtOAc 9:1) to afford 12 (0.035 g, 68%), as an oil. Rf 0.30 (hexane/EtOAc 9:1); [α]D +4.56 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.21 (m, 18H, aromatic), 7.07–7.05 (m, 2H, aromatic), 5.25 (s, 1H, H-1), 5.08 (d, J = 11.6 Hz, 1H, PhCH2), 4.80 (d, J = 11.6 Hz, 1H, PhCH2), 4.72 (d, J = 12.4 Hz, 1H, PhCH2), 4.60 (d, J = 12.4 Hz, 1H, PhCH2), 4.45 (d, J = 12.4 Hz, 2H, PhCH2), 4.32 (d, J = 11.6 Hz, 1H, PhCH2), 4.17 (d, J = 11.6 Hz, 1H, PhCH2), 4.14–4.10 (band, 2H, H-4 and H-6), 3.68 (dd, J = 8.8, 2.0 Hz, 1H, H-5), 3.60 (dd, J = 6.0, 3.0 Hz, 1H, H-7a), 3.52–3.49 (br, 1H, H-7b), 3.48 (s, 3H, OMe), 2.37 (t, J = 7.2 Hz, 2H, -C≡CCH2-), 1.55–1.51 (m, 1H, -C≡CCH2CH2-), 1.42–1.35 (m, 1H, -C≡CCH2CH2-), 1.30–1.18 (m, 6H, -C≡C(CH2)2(CH2)3-), 0.86 (t, J = 6.8 Hz, 3H, -C≡C(CH2)5CH3); 13C NMR (100 MHz, CDCl3) δ 160.9 (C-3), 138.4–137.5 (aromatic), 128.4–127.4 (aromatic), 109.9 (C-2), 100.8 (C-1), 97.4 (-C≡CCH2CH2-), 80.4 (C-5), 78.6 (C-4), 75.7 (-C≡CCH2CH2-), 73.3 (PhCH2), 72.8 (PhCH2), 71.8 (PhCH2), 71.2 (PhCH2), 71.1 (C-6), 70.8 (C-7), 55.9 (OMe), 31.3 (-C≡C(CH2)3CH2-), 28.6 (-C≡C(CH2)4CH2-), 28.5 (-C≡CCH2CH2-), 22.5 (-C≡C(CH2)2CH2-), 20.0 (-C≡CCH2(CH2)4CH3), 14.3 (-C≡C(CH2)5CH3); HRMS–ESI (m/z): [M + Na]+ calcd for 697.3505; found, 697.3507.
Methyl 2-deoxy-2-C-(2-(tert-butoxycarbonyl)ethyl)-α-D-manno-sept-3-uloside (13): A mixture of 4 (0.038 g, 0.054 mmol) and Pd/C (10%, 0.030 g) in MeOH (10 mL) was stirred under a positive pressure of H2 for 24 h at rt, filtered through a celite pad, and washed with MeOH (2 × 15 mL), and the solvents were removed in vacuo to afford 13 (0.017 g, 94%), as an oil. Rf 0.3 (MeOH/CHCl3 1:1); [α]D +63.12 (c 0.5, MeOH); 1H NMR (400 MHz, CD3OD) δ 4.37 (d, J = 8.0 Hz, 1H, H-1), 4.28 (app. d, J = 7.6 Hz, 1H, H-4), 4.08 (m, 1H, H-6), 3.85 (dd, J = 13.6, 2.4 Hz, 1H, H-7a), 3.73 (dd, J = 13.6, 4.8 Hz, 1H, H-7b), 3.46 (s, 3H, OMe), 3.35 (br, 1H, H-5), 3.22–3.17 (m, 1H, H-2), 2.21 (t, J = 7.4 Hz, 2H, -CH2-CH2CO2t-Bu), 2.10–2.01 (m, 1H, -CHHCH2CO2t-Bu), 1.83–1.75 (m, 1H, -CHHCH2CO2t-Bu), 1.49 (s, 9H, t-Bu); 13C NMR (100 MHz, CD3OD) δ 208.4 (C-3), 175.3 (C=O), 103.4 (C-1), 84.5 (C-4), 83.1 (C-t-Bu), 73.2 (C-5), 71.0 (C-6), 62.7 (C-7), 56.6 (OMe), 52.2 (C-2), 33.9 (-CH2CH2CO2t-Bu), 28.2 (t-Bu), 23.3 (-CH2CH2CO2t-Bu); HRMS–ESI (m/z): [M + Na]+ calcd for 357.1525; found, 357.1526.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectroscopic data. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Collins, P. M.; Ferrier, R. J. Monosaccharides: Their Chemistry and Their Roles in Natural Products. John Wiley & Sons: Chichester, UK, 1998; pp 40–42.
Return to citation in text: [1] -
Pakulski, Z. Pol. J. Chem. 2006, 80, 1293–1326.
Return to citation in text: [1] -
Stevens, J. D. J. Chem. Soc. D 1969, 1140–1141. doi:10.1039/C29690001140
Return to citation in text: [1] -
Stevens, J. D. Carbohydr. Res. 1972, 21, 490–492. doi:10.1016/S0008-6215(00)84936-8
Return to citation in text: [1] -
Anet, E. F. L. J. Carbohydr. Res. 1968, 8, 164–174. doi:10.1016/S0008-6215(00)80152-4
Return to citation in text: [1] -
Contour, M.-O.; Fayet, C.; Gelas, J. Carbohydr. Res. 1990, 201, 150–152. doi:10.1016/0008-6215(90)84232-J
Return to citation in text: [1] -
Tauss, A.; Steiner, A. J.; Stütz, A. E.; Tarling, C. A.; Withers, S. G.; Wrodnigg, T. M. Tetrahedron: Asymmetry 2006, 17, 234–239. doi:10.1016/j.tetasy.2005.12.007
Return to citation in text: [1] -
Sizun, G.; Dukhan, D.; Griffon, J.-F.; Griffe, L.; Meillon, J.-C.; Leroy, F.; Storer, R.; Sommadossi, J.-P.; Gosselin, G. Carbohydr. Res. 2009, 344, 448–453. doi:10.1016/j.carres.2008.12.019
Return to citation in text: [1] -
Butcher, M. E.; Ireson, J. C.; Lee, J. B.; Tyler, M. J. Tetrahedron 1977, 33, 1501–1507. doi:10.1016/0040-4020(77)88012-5
Return to citation in text: [1] -
Ovaa, H.; Leeuwenburgh, M. A.; Overkleeft, H. S.; van der Marel, G. A.; van Boom, J. H. Tetrahedron Lett. 1998, 39, 3025–3028. doi:10.1016/S0040-4039(98)00324-4
Return to citation in text: [1] -
Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9
Return to citation in text: [1] -
Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567
Return to citation in text: [1] -
Alcázar, E.; Pletcher, J. M.; McDonald, F. E. Org. Lett. 2004, 6, 3877–3880. doi:10.1021/ol0483495
Return to citation in text: [1] -
Hoberg, J. O.; Bozell, J. J. Tetrahedron Lett. 1995, 36, 6831–6834. doi:10.1016/0040-4039(95)01387-W
Return to citation in text: [1] -
Cousins, G. S.; Hoberg, J. O. Chem. Soc. Rev. 2000, 29, 165–174. doi:10.1039/a906932a
Return to citation in text: [1] -
Ramana, C. V.; Murali, R.; Nagarajan, M. J. Org. Chem. 1997, 62, 7694–7703. doi:10.1021/jo970948k
Return to citation in text: [1] -
Hewitt, R. J.; Harvey, J. E. J. Org. Chem. 2010, 75, 955–958. doi:10.1021/jo902306a
Return to citation in text: [1] -
Fukami, H.; Koh, H.-S.; Sakata, T.; Nakajima, M. Tetrahedron Lett. 1968, 9, 1701–1704. doi:10.1016/S0040-4039(01)99032-X
Return to citation in text: [1] -
Wang, Z.-X.; Miller, S. M.; Anderson, O. P.; Shi, Y. J. Org. Chem. 1999, 64, 6443–6458. doi:10.1021/jo9908849
Return to citation in text: [1] -
Köver, A.; Matheu, M. I.; Díaz, Y.; Castillón, S. ARKIVOC 2007, part (iv), 364–379.
Return to citation in text: [1] -
Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2009, 74, 739–746. doi:10.1021/jo801967s
Return to citation in text: [1] -
Ganesh, N. V.; Raghothama, S.; Sonti, R.; Jayaraman, N. J. Org. Chem. 2010, 75, 215–218. doi:10.1021/jo901945e
Return to citation in text: [1] -
Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024
Return to citation in text: [1] -
de Meijere, A.; Meyer, F. E. Angew. Chem. 1994, 106, 2473–2506. doi:10.1002/ange.19941062307
Return to citation in text: [1] -
Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/S0040-4039(01)95429-2
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/S0040-4039(00)91094-3
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e
Return to citation in text: [1] -
Al-Harrasi, A.; Fischer, S.; Zimmer, R.; Reissig, H.-U. Synthesis 2010, 304–314. doi:10.1055/s-0029-1217126
Return to citation in text: [1]
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 1. | Collins, P. M.; Ferrier, R. J. Monosaccharides: Their Chemistry and Their Roles in Natural Products. John Wiley & Sons: Chichester, UK, 1998; pp 40–42. |
| 9. | Butcher, M. E.; Ireson, J. C.; Lee, J. B.; Tyler, M. J. Tetrahedron 1977, 33, 1501–1507. doi:10.1016/0040-4020(77)88012-5 |
| 10. | Ovaa, H.; Leeuwenburgh, M. A.; Overkleeft, H. S.; van der Marel, G. A.; van Boom, J. H. Tetrahedron Lett. 1998, 39, 3025–3028. doi:10.1016/S0040-4039(98)00324-4 |
| 28. | Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 16, 4467–4470. doi:10.1016/S0040-4039(00)91094-3 |
| 29. | Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e |
| 4. | Stevens, J. D. Carbohydr. Res. 1972, 21, 490–492. doi:10.1016/S0008-6215(00)84936-8 |
| 5. | Anet, E. F. L. J. Carbohydr. Res. 1968, 8, 164–174. doi:10.1016/S0008-6215(00)80152-4 |
| 6. | Contour, M.-O.; Fayet, C.; Gelas, J. Carbohydr. Res. 1990, 201, 150–152. doi:10.1016/0008-6215(90)84232-J |
| 7. | Tauss, A.; Steiner, A. J.; Stütz, A. E.; Tarling, C. A.; Withers, S. G.; Wrodnigg, T. M. Tetrahedron: Asymmetry 2006, 17, 234–239. doi:10.1016/j.tetasy.2005.12.007 |
| 8. | Sizun, G.; Dukhan, D.; Griffon, J.-F.; Griffe, L.; Meillon, J.-C.; Leroy, F.; Storer, R.; Sommadossi, J.-P.; Gosselin, G. Carbohydr. Res. 2009, 344, 448–453. doi:10.1016/j.carres.2008.12.019 |
| 30. | Al-Harrasi, A.; Fischer, S.; Zimmer, R.; Reissig, H.-U. Synthesis 2010, 304–314. doi:10.1055/s-0029-1217126 |
| 24. | Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320–2322. doi:10.1021/jo00979a024 |
| 25. | de Meijere, A.; Meyer, F. E. Angew. Chem. 1994, 106, 2473–2506. doi:10.1002/ange.19941062307 |
| 26. | Miyaura, N.; Yamada, K.; Suzuki, A. Tetrahedron Lett. 1979, 20, 3437–3440. doi:10.1016/S0040-4039(01)95429-2 |
| 27. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 20. | Köver, A.; Matheu, M. I.; Díaz, Y.; Castillón, S. ARKIVOC 2007, part (iv), 364–379. |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 18. | Fukami, H.; Koh, H.-S.; Sakata, T.; Nakajima, M. Tetrahedron Lett. 1968, 9, 1701–1704. doi:10.1016/S0040-4039(01)99032-X |
| 19. | Wang, Z.-X.; Miller, S. M.; Anderson, O. P.; Shi, Y. J. Org. Chem. 1999, 64, 6443–6458. doi:10.1021/jo9908849 |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 14. | Hoberg, J. O.; Bozell, J. J. Tetrahedron Lett. 1995, 36, 6831–6834. doi:10.1016/0040-4039(95)01387-W |
| 15. | Cousins, G. S.; Hoberg, J. O. Chem. Soc. Rev. 2000, 29, 165–174. doi:10.1039/a906932a |
| 16. | Ramana, C. V.; Murali, R.; Nagarajan, M. J. Org. Chem. 1997, 62, 7694–7703. doi:10.1021/jo970948k |
| 17. | Hewitt, R. J.; Harvey, J. E. J. Org. Chem. 2010, 75, 955–958. doi:10.1021/jo902306a |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 11. | Peczuh, M. W.; Snyder, N. L. Tetrahedron Lett. 2003, 44, 4057–4061. doi:10.1016/S0040-4039(03)00849-9 |
| 12. | Schmidt, B.; Biernat, A. Chem.–Eur. J. 2008, 14, 6135–6141. doi:10.1002/chem.200800567 |
| 13. | Alcázar, E.; Pletcher, J. M.; McDonald, F. E. Org. Lett. 2004, 6, 3877–3880. doi:10.1021/ol0483495 |
| 21. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2007, 72, 5500–5504. doi:10.1021/jo070444e |
| 22. | Ganesh, N. V.; Jayaraman, N. J. Org. Chem. 2009, 74, 739–746. doi:10.1021/jo801967s |
| 23. | Ganesh, N. V.; Raghothama, S.; Sonti, R.; Jayaraman, N. J. Org. Chem. 2010, 75, 215–218. doi:10.1021/jo901945e |
© 2012 Dey and Jayaraman; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)