Abstract
Mycoplasma fermentans possesses unique α-glycolipid antigens (GGPL-I and GGPL-III) at the cytoplasm membrane, which carry a phosphocholine group at the sugar primary (6-OH) position. This paper describes a practical synthetic pathway to a GGPL-I homologue (C16:0) and its diastereomer, in which our one-pot α-glycosylation method was effectively applied. The synthetic GGPL-I isomers were characterized with 1H NMR spectroscopy to determine the equilibrium among the three conformers (gg, gt, tg) at the acyclic glycerol moiety. The natural GGPL-I isomer was found to prefer gt (54%) and gg (39%) conformers around the lipid tail, while adopting all of the three conformers with equal probability around the sugar position. This property was very close to what we have observed with respect to the conformation of phosphatidylcholine (DPPC), suggesting that the Mycoplasma glycolipids GGPLs may constitute the cytoplasm fluid membrane together with ubiquitous phospholipids, without inducing stereochemical stress.

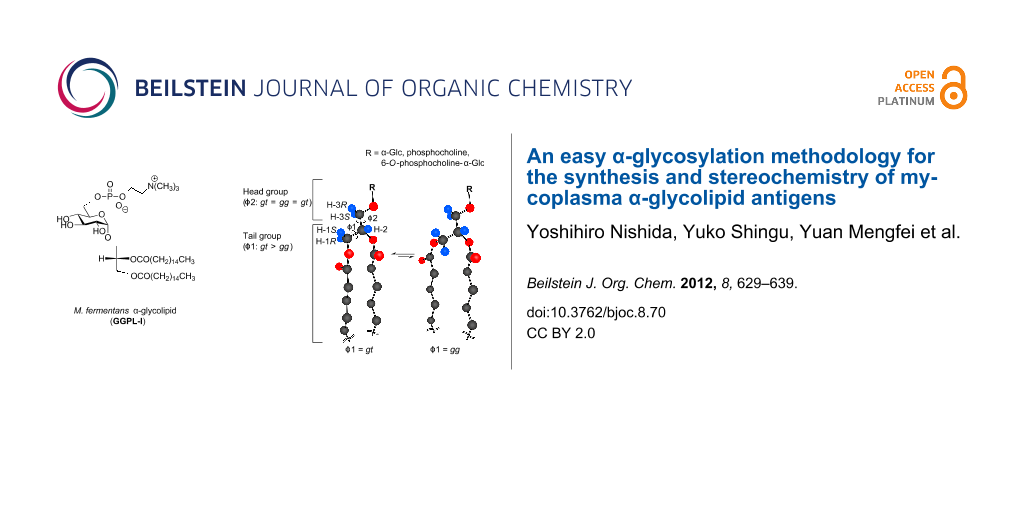
Graphical Abstract
Introduction
Mycoplasmas constitute a family of gram-positive microbes lacking rigid cell walls. They are suspected to be associated with human immune diseases, in either direct or indirect ways, although the molecular mechanism is not fully understood [1]. In recent biochemical studies, Mycoplasma outer-membrane lipoproteins [2,3] and glycolipids [4-6] are thought to serve not only as the main antigens but also as probable pathogens. Also in our research team, Matsuda et al. [7-10] isolated a new class of α-glycolipid antigens (GGPL-I and GGPL-III, Figure 1) from M. fermentans. Another α-glycolipid (MfGL-II), which has a chemical structure very close to GGPL-III, was identified and characterized by other groups [11-14].
![[1860-5397-8-70-1]](/bjoc/content/figures/1860-5397-8-70-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Absolute chemical structures of M. fermentans α-glycolipid antigens, GGPL-I and GGPl-III (GGPL: Glycosyl-sn-glycerophospholipid).
Figure 1: Absolute chemical structures of M. fermentans α-glycolipid antigens, GGPL-I and GGPl-III (GGPL: Gly...
Absolute chemical structures of GGPL-I [15] and GGPL–III [16] have already been established by chemical syntheses of stereoisomers; these α-glycolipids have a common chemical backbone of 3-O-(α-D-glucopyranosyl)-sn-glycerol carrying phosphocholine at the sugar primary (6-OH) position. The fatty acids at the glycerol moiety are saturated, namely palmitic acid (C16:0) and stearic acid (C18:0). GGPL-I has a structural feature analogous to 1,2-di-O-palmitoyl phosphatidylcholine (DPPC) as a ubiquitous cell membrane phospholipid. Apparently, GGPLs are amphiphilic compounds that can form certain self-assembled structures under physiological conditions [12,13] and may give physicochemical stress on the immune system of the host [17]. In fact, our research team has proven that these α-glycolipid antigens have certain pathogenic functions [18,19].
In order to exploit their biological functions in detail, it is necessary to obtain these α-glycolipids in sufficient amounts. Thus, both genetic [20-22] and chemical synthetic approaches [23,24] are being followed, although no practical way has yet been established. In this paper, we report a chemical access to both a natural GGPL-I homologue (C16:0) and its diastereomer I-a and I-b, in which our one-pot α-glycosylation methodology [25,26] is effectively applied. The two GGPL-I isomers prepared thereby were characterized with 1H NMR spectroscopy, in terms of configuration and conformation at the asymmetric glycerol moiety.
Results and Discussion
A practical synthetic access to GGPL-I homologues
GGPL-I provides two key asymmetric centers to be controlled, literally, in the synthetic pathway. One is the configuration at the chiral glycerol moiety, and another is the sugar α-glycoside linkage. In former synthetic works on 3-O-(α-D-glycopyranosyl)-sn-glycerol [27-30], chiral 1,2-O-isopropylidene-sn-glycerol has often been employed [29,30] as the acceptor substrate for different α-glycosylation reactions. In this case, however, attention should be paid to the acid-catalyzed migration of the dimethylketal group [23,29-31]. In our synthetic pathway, chiral (S)- or (R)-glycidol is employed as an alternative source of the chiral glycerol to circumvent this problem. In an established synthetic approach, 6-O-acetyl-2,3,4-tri-O-benzyl protected sugar 1 [23] is used as the donor and treated with a reagent combination of CBr4 and Ph3P (Appel–Lee reagent) in either CH2Cl2 or DMF solvent, or a mixture of the two (Scheme 1). For the reaction in CH2Cl2, N,N,N’,N’-tetramethylurea (TMU) is added after in situ formation of α-glycosyl bromide 2, which equilibrates with a more reactive β-glycosyl bromide species [32]. In the pathway using DMF, the α-glycosylation is routed via α-glycosyl cationic imidate 3, which was predicted in former studies [33] and evidenced in our preceding NMR and MS study [25,26].
![[1860-5397-8-70-i1]](/bjoc/content/inline/1860-5397-8-70-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: An established synthetic pathway to α-glycosyl-sn-glycerols 4a and 5a. A reagent combination of CBr4 and Ph3P (Appel–Lee reagent) is utilized in either CH2Cl2 or DMF as solvent.
Scheme 1: An established synthetic pathway to α-glycosyl-sn-glycerols 4a and 5a. A reagent combination of CBr4...
The reaction between 1 and (S)-glycidol in CH2Cl2 (+ TMU) gave a mixture of epoxy compound 4a (60–70%) and bromide 5a (30–40%). In 5a, the oxirane ring was opened by nucleophilic Br− ions produced by Ph3P and CBr4. Also in the DMF-promoted reaction, a mixture of 4a (70–90%) and 5a (10–30%) was derived. In both reaction pathways, however, the glycosylation was α-selective (α:β ≥ 90:10, yields >80%) and not accompanied by isomerization at the glycerol moiety.
A mixture of 4a and 5a was used in the following chemical transformation (Scheme 2). First, a lyso-glycolipid 6a was derived after deprotection at the sugar hydroxymethyl position and SN2 substitution with cesium palmitate at the glycerol sn-1 position. Then, this compound was converted to glycolipid 8a after sequential reaction of the temporary tert-butyldimethylsilyl (TBDMS) -protected sugar, and O-acylation at the glycerol 2-OH position to give 7a, followed by removal of the TBDMS protecting group. For introducing the phosphocholine group at the sugar 6-OH position, we employed a phosphoroamidite method using 1H-tetrazole as a promoter [34]. First, 8a was treated with 2-cyanoethyl-N,N,N’,N’-tetraisopropyl phosphorodiamidite in the presence of 1H-tetrazole, and then with choline tosylate to give 9a. After removal of the sugar O-benzyl group by catalytic hydrogenolysis, the GGPL-I homologue I-a was obtained. In the same way, the GGPL-I sn-isomer I-b was derived from a mixture of 4b and 5b available from the reaction between 1 and (R)-glycidol (Scheme 1 and Scheme 2b).
![[1860-5397-8-70-i2]](/bjoc/content/inline/1860-5397-8-70-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of GGPL-I homologue I-a and its isomer I-b. Conditions: (a) K2CO3, CH3OH; (b) cesium palmitate in DMF; (c) TBDMS chloride then palmitoyl chloride in pyridine + DMAP; (d) TFA in CH3OH; (e) (i) 2-cyanoethyl-N,N,N’,N’-tetraisopropyl phosphorodiamidite, 1H-tetrazole and MS-4 Å in CH2Cl2; (ii) choline tosylate, 1H-tetrazole, (iii) mCPBA, (iv) aq. NH3 in CH3OH, (f) H2, Pd(OH)2/C in CH3OH.
Scheme 2: Syntheses of GGPL-I homologue I-a and its isomer I-b. Conditions: (a) K2CO3, CH3OH; (b) cesium palm...
1H NMR characterization of I-a, I-b and the related glycerolipids
1H NMR spectroscopy provides a useful tool for discriminating between the two GGPL-I isomers as shown in Figure 2. A clear difference was observed in the chemical shifts of the glycerol methylene protons as designated by “a” and “b”. Conversely, little difference was observed between the sn-isomers at the sugar H-1 signal as well as at the glycerol H-2 (Table 1). Natural GGPL-I and GGPL-III gave 1H NMR data very close to those of I-a, indicating that both have a common skeleton of 3-O-(α-D-glucopyranosyl)-sn-glycerol [15,16].
![[1860-5397-8-70-2]](/bjoc/content/figures/1860-5397-8-70-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectra of I-a and I-b (500 MHz, 25 °C, CDCl3/CD3OD 10:1). The assignment of sn-glycerol methylene protons (HproR and HproS) was performed on the basis of our preceding studies on deuterium-labeled glycerols [35-37] and α(1→6)-linked disaccharides [38-40].
Figure 2: 1H NMR spectra of I-a and I-b (500 MHz, 25 °C, CDCl3/CD3OD 10:1). The assignment of sn-glycerol met...
Table 1: 1H NMR data (500 MHz) of I-a, I-b, and their precursors (9a and 9b).
| δ [ppm] ( 3J and 2J [Hz] ) | ||||||
|---|---|---|---|---|---|---|
| Compound | glucose | sn-glycerol moiety | ||||
| H-1’ | H-1 | H-2 | H-3 | |||
| proR | proS | proR | proS | |||
| I-aa | 4.80 (3.5) | 4.14 (6.5) | 4.40 (3.5, 12.0) | 5.24 | 3.76 (5.5) | 3.61 (5.5, 11.0) |
| 9a | 4.70 (3.5) | 4.17 (6.5) | 4.38 (3.0, 12.0) | 5.22 | 3.72 (5.5) | 3.52 (6.0, 11.0) |
| I-ba | 4.80 (3.5) | 3.80 (6.0) | 3.57 (5.5,11.0) | 5.23 | 4.34 (3.5) | 4.20 (6.5,12.0) |
| 9b | 4.74 (3.5) | 3.76 (5.5) | 3.57 (5.5, 11.0) | 5.23 | 4.37 (3.0) | 4.19 (6.5, 12.0) |
aThese α-glycolipids were dissolved in a mixture of CDCl3 and CD3OD (10:1) at 11.2 mM concentration.
The glycerol moiety has two C–C single bonds. By free rotation, each of them is allowed to have three staggered conformers of gg (gauche-gauche), gt (gauche-trans) and tg (trans-gauche) (Figure 3). In solution and also in self-contacting liquid-crystalline states, these conformers are thought to equilibrate with each other. In this study, we calculated time-averaged populations of the three conformers by means of 1H NMR spectroscopy. As we reported in a preceding paper [41], the Karplus-type equation proposed by Haasnoot et al.[42] was adapted as follows:
2.8gg + 3.1gt + 10.7tg = 3JH2,H1S (or 3JH2,H3R)
0.9gg + 10.7gt + 5.0tg = 3JH2,H1R (or 3JH2,H3S) and
gg + gt + tg = 1.
In this equation, a perfect staggering (Φ1 and Φ2 = +60, −60 or 180 degree) is assumed for every conformer. Figure 3 summarizes the results for a series of 3-substituted 1,2-di-O-palmitoyl-sn-glycerols, which involve tripalmitin (Figure 3, entry 1), DPPC (1,2-di-O-palmitoylphosphatidylcholine) (Figure 3, entry 2), and GGPL-I homologues (Figure 3, entries 3–5). In a solution state with a mixture of CDCl3 and CD3OD (10:1) as the solvent and at a concentration of 11.2 mM, tripalmitin adopts the three conformers in the ratio of gt (45%), gg (37%) and tg (18%). In comparison with this symmetric lipid, the asymmetric phospholipid (DPPC) favors the gt-conformer more strongly around the tail lipid moiety along the sn-1,2 position, while disfavoring the tg-conformer, in the ratio of gt (59%), gg (34%) and tg (7%). The head phosphate moiety along the sn-2,3 position adopts the three conformers in equilibrated populations (gg = gt = tg).
![[1860-5397-8-70-3]](/bjoc/content/figures/1860-5397-8-70-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Distributions of gg, gt and tg-conformers in 3-substituted sn-glycerols at 11 mM in solutions of CDCl3 and CD3OD (10:1) at 298 K.
Figure 3: Distributions of gg, gt and tg-conformers in 3-substituted sn-glycerols at 11 mM in solutions of CD...
3-O-(α-D-glucopyranosyl)-sn-glycerolipids 10 and 11 (Figure 3, entries 3 and 4) were found to have conformational properties very similar to DPPC; the lipid tail moiety prefers the gauche conformations (gt and gg), while the sugar moiety allows a random conformation. Here, it should be mentioned that the conformer distribution coincides between I-a (Figure 3, entry 5) and DPPC (Figure 3, entry 2) at the head moiety [gt (33%), gg (34%) and tg (33%)].
The above analysis was carried out also for the stereoisomer I-b and the related glycolipids (Figure 4, entries 6–8). The isomer (Figure 4, entry 8) showed an overall conformational property similar to I-a and DPPC (Figure 5), although a small difference was observed in the conformer distribution at the sugar head moiety. However, it should be recognized here that the helical direction (helicity) of the gt conformer in I-b is reversed (anticlockwise) from the case of DPPC and GGPL-I (clockwise), as depicted in Figure 3 and Figure 4.
![[1860-5397-8-70-4]](/bjoc/content/figures/1860-5397-8-70-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Distributions of gg, gt and tg-conformers in 1-substituted sn-glycerols. In these sn-isomers, Φ1 and Φ2 represent the dihedral angles around the C–C single bond at the glycerol sn-2,3 and 1,2-position, respectively.
Figure 4: Distributions of gg, gt and tg-conformers in 1-substituted sn-glycerols. In these sn-isomers, Φ1 an...
![[1860-5397-8-70-5]](/bjoc/content/figures/1860-5397-8-70-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: A common conformational property of GGPL-I and DPPC. The tail lipid moiety favors two gauche-conformers of gt and gg (gt > gg >> tg), while the head moiety takes three conformers in averaged populations (gt = gg = tg).
Figure 5: A common conformational property of GGPL-I and DPPC. The tail lipid moiety favors two gauche-confor...
Conclusion
We have proposed a synthetic pathway to a GGPL-I homologue and its stereoisomer, in which our one-pot α-glycosylation methodology was effectively applied. We envisage that the simple method will allow us to prepare a variety of α-glycolipid antigens other than GGPLs and to prove their biological significance [43]. By the 1H NMR conformational analysis, which was based on our former studies on deuterium-labeled sn-glycerols and sugars, we have proven that GGPL-I and other 3-O-(α-D-glucopyranosyl)-sn-glycerolipids have a common conformational property at the chiral glycerol moiety: The lipid tail moiety prefers two gauche-conformations (gg and gt) in the order gt > gg >> tg, while the sugar head moiety adopts three conformers in an averaged population (gg = gt = tg). At the lipid tail position, the gt-conformer with clockwise helicity is predominant over the anticlockwise gg-conformer. The observed conformation was very close to what we have seen in DPPC (Figure 5). Although these results were based on the solution state in a solvent mixture of CHCl3 and CH3OH (10:1), it may be possible to assume that the mycoplasma GGPLs and the related 3-O-(α-D-glycopyranosyl)-sn-glycerolipids can constitute cytoplasm membranes in good cooperation with ubiquitous phospholipids without inducing stereochemical stress at the membrane.
The GGPL-I isomer I-b showed an overall conformational property similar to the natural isomer I-a and DPPC. However, it should be mentioned here that the chiral helicity of gt-conformers in I-b is reversed (anticlockwise) from the clockwise helicity of DPPC and GGPL-I. The difference in chirality seems critical in biological recognition events and also in physicochemical contact with other chiral constituents in cell membranes [44,45].
Experimental
General methods
Infrared (IR) spectra were recorded on a JASCO FT/IR-230 Fourier transform infrared spectrometer on KBr disks. All 1H NMR (500 MHz) spectra were recorded by using a Varian INOVA-500 or Varian Gemini 200. 1H chemical shifts are expressed in parts per million (δ ppm) by using an internal standard of tetramethylsilane (TMS = 0.000 ppm). Mass spectra were recorded with a JEOL JMS 700 spectrometer for fast atom bombardment (FAB) spectra. Silica gel column chromatography was performed on silica gel 60 (Merck 0.063–0.200 mm and 0.040–0.063 mm) and eluted with a mixture of toluene and ethyl acetate or a mixture of CHCl3 and CH3OH in gradient modes (100:0 to 80:20). For purification of phosphocholine-containing products, a chromatographic column packed with Iatrobeads (IATRON LABORATORIES INC., 6RS-8060) was applied and eluted with a mixture of CH3OH and CHCl3 in gradient modes. For thin-layer chromatography (TLC) analysis, Merck precoated TLC plates (silica gel 60 F254, layer thickness 0.25 mm) and Merck TLC aluminum roles (silica gel 60 F254, layer thickness 0.2 mm) were used. All other chemicals were purchased from Tokyo Kasei Kogyo Co., Ltd., Kishida Chemical Co., Ltd., and Sigma–Aldrich Chemical Company Co, Int., and were used without further purification.
A typical procedure for the one-pot α-glycosylation: CBr4 (1.6 g, 6.09 mmol) and Ph3P (2.02 g, 6.09 mmol) were added to a solution of 6-O-acetyl-2,3,4-tri-O-benzyl-D-glucose (1) (1.0 g, 2.03 mmol) in 10 mL of DMF and stirred for 3 h at rt. Then, (S)-glycidol (301 mg, 4.06 mmol) was added to the reaction mixture and stirred for 14 h at rt. Products were diluted with a mixture of toluene and ethyl acetate (10:1), and the solution was washed with saturated aq. NaHCO3 and aq. NaCl solution, dried and concentrated. The residue was purified by silica gel column chromatography in toluene and ethyl acetate to give a mixture of 4a and 5a (the ratio changed with reaction time) as colorless syrup. The total yield of 4a and 5a was between 80% and 90%.
3-O-(2,3,4-tri-O-benzyl-α-D-glucopyranosyl)-1,2-di-O-palmitoyl-sn-glycerols 8a and 8b: K2CO3 (379 mg, 2.74 mmol) was added to the mixture of 4a and 5a (1 g, 1.83 mmol based on 4a) in CH3OH (20 mL) and stirred for 1 h at rt. The reaction mixture was neutralized, washed with water, dried, and concentrated. The residue was dried under reduced pressure and directly subjected to the next reaction. A mixture of caesium palmitate (2.7 g, 7.3 mmol) in DMF (40 mL) was heated at 100–110 °C, to which the DMF solution of the above residue was added slowly. The reaction mixture was stirred for 2 h at 110 °C, cooled to rt, and then filtered through a pad of Celite powder with ethyl acetate. The filtrate was washed with saturated aq. NaCl solution, dried, and concentrated. The residue was purified by silica gel column chromatography to give 6a as a colorless syrup (830 mg, 60% yield). To a solution of 6a (300 mg, 0.39 mmol) in pyridine (20 mL), TBDMS chloride (107 mg, 0.71 mmol) and 4-N,N-dimethylaminopyridine (cat.) were added. The reaction mixture was stirred for 12 h at rt, treated with methanol (2 mL) for 3 h and concentrated. The residue was purified by silica gel column chromatography in a mixture of toluene and ethyl acetate. The main product was dissolved in pyridine (20 mL) and then reacted with palmitoyl chloride (162 mg, 0.59 mmol) for 3 h at rt. The reaction mixture was treated with methanol (2 mL) and then concentrated with toluene. The residue was dissolved in a mixture of CH3OH and CH2Cl2 (1:1, 20 mL) and treated with trifluoroacetic acid (1 mL) for 2 h at rt. After concentration, the residue was purified by silica gel column chromatography in a mixture of toluene and ethyl acetate to give 8a as a white waxy solid (0.32 g, 81% yield from 6a). [α]D30 +21.1 (c 1.0, CHCl3); IR (KBr, film): 3413, 2923, 2853, 1736, 1630, 1457, 1361, 1158, 1069, 736 cm−1; 1H NMR (500 MHz, CDCl3): δH 7.40–7.23 (m, 5H × 3, -CH2C6H5), 5.23 (m, 1H, glycerol H-2), 4.96–4.64 (d, 2H × 3,-CH2C6H5), 4.70 (d, 1H, J = 3.5 Hz, H-1), 4.40 (dd, 1H, J = 4.0 and 12.0 Hz, glycerol H-1proS), 4.19 (dd, 1H, J = 6.0 and 12.0 Hz, glycerol H-1proR), 3.96 (dd, 1H, J = 9.5 and 9.5 Hz, H-3), 3.72 (dd, 1H, J = 5.5 and 10.5 Hz, glycerol H-3proR), 3.72 and 3.66 (b, 2H, H-6ProR and H-6proS), 3.65 (m, 1H, H-5), 3.54 (dd, 1H, J = 5.5 and 10.5 Hz, glycerol H-3proS), 3.50 (dd, 1H, J = 9.5 and 10.0 Hz, H-4), 3.49 (dd, 1H, J = 3.5 and 9.5 Hz, H-2), 2.29 (m, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.58 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.25 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); FABMS m/z: [M + Na]+ 1023.7.
In the same way as derived for the synthesis of 8a, (R)-glycidol and 6b (0.30 g, 0.39 mmol) was used for the synthesis of 8b (0.30 g, 77% yield). [α]D32 +18.5 (c 1.0, CHCl3); IR (KBr, film): 3452, 2924, 2854, 1739, 1586, 1455, 1296, 1159, 1095, 710 cm−1; 1H NMR (500 MHz, CDCl3): δH 7.40–7.23 (m, 5H × 3, -CH2C6H5), 5.23 (b, 1H, glycerol H-2), 4.96–4.63 (d, 2H × 3,-CH2C6H5), 4.75 (d, 1H, J = 3.5 Hz, H-1), 4.38 (dd, 1H, J = 3.5 and 12.0 Hz, glycerol H-3proR), 4.21 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-3proS), 3.96 (dd, 1H, J = 9.5 and 9.5 Hz, H-3), 3.73 (dd, 1H, J = 6.0 and 11.0 Hz, glycerol H-1proS), 3.76 and 3.66 (b, 2H, H-6ProR and H-6proS), 3.65 (m, 1H, H-5), 3.57 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-1proR), 3.51 (dd, 1H, J = 9.5 and 10.0 Hz, H-4), 3.50 (dd, 1H, J = 3.5 and 9.5 Hz, H-2), 2.29 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.59 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.25 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); FABMS m/z: [M + Na]+ 1023.7.
A phosphorodiamidite method for the synthesis of I-a
(a) The reaction vessel was kept under anhydrous conditions with Ar gas in the presence of molecular sieves (50%w/w), and a solution of 8a (0.20 g, 0.20 mmol) and 2-cyanoethyl-N,N,N',N'-tetraisopropyl phosphorodiamidite (90.4 mg, 0.30 mmol) in 10 mL of CH2Cl2 was injected. 1H-tetrazole (28.4 mg, 0.40 mmol) was added and stirred for 2 h at rt. Then 1H-tetrazole (42.6 mg, 0.60 mmol, 3.0 equiv) and choline tosylate (220.3 mg, 0.8 mmol: thoroughly dried overnight under vacuum) were added to the reaction mixture and stirred for 1.5 h at rt. The reaction was quenched by the addition of water (1 mL), and then m-chloroperbenzoic acid (51.8 mg, 0.3 mmol) was added at 0 °C and stirred for 10 min at rt. The reaction mixture was washed with 10 % aq. Na2SO3 solution, saturated aq. NaHCO3 solution, water and saturated aq. NaCl solution, dried and concentrated. The residue was dissolved in a mixture of CH3OH (10 mL) and 30% aq. NH3 (1 mL) and stirred for 15 min at rt. The reaction mixture was concentrated, and the residue was purified by column chromatography (IATROBEADS in a mixture of CHCl3 and CH3OH) to give 9a (186 mg, 80% yield). [α]D26 +13.0 (c 0.45, CHCl3); IR (KBr, film): 3301, 2929, 2856, 2537, 1731, 1577, 1419, 1216, 1093, 925, 788, 746; 1H NMR (500 MHz, CDCl3): δH 7.35–7.25 (m, 5H × 3, -CH2C6H5), 5.22 (m, 1H, glycerol H-2), 4.93–4.61 (d, 2H × 3, -CH2C6H5), 4.70 (d, 1H, J = 3.5 Hz, H-1), 4.38 (dd, 1H, J = 3.0 and 12.0 Hz, glycerol H-1proS), 4.19 (b, 2H, choline -CH2CH2N+(CH3)3), 4.17 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-1proR), 4.15 and 4.02 (m, 2H × 2, H-6ProR and H-6proS), 3.93 (dd, 1H, J = 9.0 and 9.5 Hz, H-3), 3.72 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-3proR), 3.71 (b, 1H, H-5), 3.62 (t, 1H, H-4), 3.58 (b, 2H, choline -CH2CH2N+(CH3)3), 3.52 (dd, 1H, J = 6.0 and 11.0 Hz, glycerol H-3proS), 3.46 (dd, 1H, J = 3.5 and 9.5 Hz, H-2), 3.15 (s, 9H, -POCH2CH2N+(CH3)3), 2.28 (m, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.58 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.25 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); FABMS m/z: [M + Na]+ 1188.7.
(b) Compound 9a (0.18 g, 0.15 mmol) was hydrogenated with Pd(OH)2/C (8 mg) under atmospheric pressure in a mixture of CH3OH (10 mL) and acetic acid (0.1 mL) for 7 h at rt. The reaction mixture was neutralized by the addition of Et3N, filtered and concentrated. The residue was purified by column chromatography with IATROBEADS (CH3OH and CHCl3) to give I-a (101 mg, 75% yield). [α]D31 +18.7 (c 1.0, CHCl3/CH3OH 10:1); IR (KBr, film): 3372, 2927, 2852, 1731, 1573, 1469, 1112, 975, 727; 1H NMR (500 MHz, CDCl3/CD3OD 10:1): δH 5.23 (m, 1H, glycerol H-2), 4.80 (d, 1H, J = 3.5 Hz, H-1), 4.39 (dd, 1H, J = 3.5 and 12.0 Hz, glycerol H-1proS), 4.30 (b, 2H, -CH2CH2N+(CH3)3), 4.24 and 3.95 (b, 2H, H-6ProR and H-6proS), 4.14 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-1proR), 3.75 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-3proR), 3.67 (b, 2H, choline-CH2CH2N+(CH3)3), 3.65 (b, 1H × 2, H-3 and H-5), 3.61 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-3proS), 3.56 (b, 1H, H-4), 3.46 (b, 1H, H-2), 3.22 (s, 9H, -POCH2CH2N+(CH3)3), 2.31 (m, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.60 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.25 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); HRMS–FAB (m/z): [M + Na]+ calcd for C46H90NO13PNa, 918.6048; found, 918.6028.
In the same way as described above, 9b (180 mg) was derived from 8b (200 mg, 0.20 mmol) in 76% yield and converted to the GGPL-I isomer I-b [70 mg, 81% yield from 9b (120 mg)]. 9b: [α]D26 +8.1 (c 0.62, CHCl3); IR (KBr, film): 3413, 2923, 2857, 1735, 1461, 1241, 1097, 744, 495, 445; 1H NMR (500 MHz, CDCl3): δH 7.35–7.23 (m, 5H × 3, -CH2C6H5), 5.23 (b, 1H, glycerol H-2), 4.94–4.64 (d, 2H × 3,-CH2C6H5), 4.74 (d, 1H, J = 3.5 Hz, H-1), 4.37 (dd, 1H, J = 3.0 and 12.0 Hz, glycerol H-3proR), 4.23 (b, 2H, choline-CH2CH2N+(CH3)3), 4.19 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-3proS), 4.16 and 4.09 (b, 2H, H-6proR and H-6proS), 3.94 (dd, 1H, J = 9.5 and 9.5 Hz, H-3), 3.76 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-1proS), 3.72 (m, 1H, H-5), 3.61 (dd, 1H, J = 9.0 and 9.5 Hz, H-4), 3.60 (b, 2H, choline-CH2CH2N+(CH3)3), 3.57 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-1proR), 3.49 (dd, 1H, J = 3.5 and 9.5 Hz, H-2), 3.20 (s, 9H, choline-CH2CH2N+(CH3)3), 2.28 (dd, 2H × 2, J = 7.5 and 15 Hz, -OCOCH2CH2(CH2)12CH3), 1.58 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.25 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); FABMS m/z: [M + Na]+ 1188.7. I-b: [α]D31= +10.7 (c 1.0, CHCl3/CH3OH 10:1); IR (KBr film): 3390, 2919, 2856, 1731, 1463, 1228, 1074, 964, 711; 1H NMR (500 MHz, CDCl3/CD3OD 10:1): δH 5.24 (m, 1H, glycerol H-2), 4.80 (d, 1H, J = 3.5 Hz, H-1), 4.34 (dd, 1H, J = 3.5 and 12.0 Hz, glycerol H-3proR), 4.28 (b, 2H, choline-CH2CH2N+(CH3)3), 4.25 and 3.97 (b, 2H, H-6ProR and H-6proS), 4.20 (dd, 1H, J = 7.0 and 12.0 Hz, glycerol H-3proS), 3.80 (dd, 1H, J = 6.0 and 11.0 Hz, glycerol H-1proS), 3.63 (b, 1H × 2, H-3 and H-5), 3.63 (b, 2H, choline-CH2CH2N+(CH3)3), 3.58 (dd, 1H, J = 5.5 and 11.0 Hz, glycerol H-1proR), 3.55 (b, 1H, H-4), 3.46 (dd, 1H, H-2), 3.22 (s, 9H, choline-CH2CH2N+(CH3)3), 2.31 (dd, 2H × 2, J = 7.5 and 15 Hz, -OCOCH2CH2(CH2)12CH3), 1.60 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.26 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); HRMS–FAB (m/z): M + Na]+ calcd for C46H90NO13PNa, 918.6048; found, 918.6078.
3-O-(α-D-glucopyranosyl)-1,2-di-O-palmitoyl-sn-glycerol (10, entry 3): Compound 10 was obtained as a waxy solid (73 mg, 83% yield) from 8a (120 mg, 0.12 mmol) by catalytic hydrogenation under the same reaction conditions as those described above for the preparation of I-a. [α]D30 +27.2 (c 1.0, CHCl3/CH3OH 10:1); IR (KBr, film): 3411, 2919, 2851, 1739, 1587, 1465, 1158, 1053, 720 cm−1; 1H NMR (500 MHz, CDCl3/CD3OD 10:1): δH 5.25 (m, 1H, glycerol H-2), 4.83 (d, 1H, J = 3.5 Hz, H-1), 4.40 (dd, 1H, J = 3.5 and 12.0 Hz, glycerol H-1proS), 4.16 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-1proR), 3.82 (dd, 1H, J = 5.5 and 10.5 Hz, glycerol H-3proR), 3.78 (d, 2H, J = 3.5 Hz, H-6proR and H-6proS), 3.65 (t, 1H, J = 9.5 and 9.5 Hz, H-3), 3.62 (dd, 1H, J = 6.0 and 10.5 Hz, glycerol H-3proS), 3.56 (dt, 1H, H-5), 3.44 (dd, 1H, J = 3.5 and 9.5 Hz, H-2), 3.42 (dd, 1H, J = 8.5 and 10.0 Hz, H-4), 2.32 (dt, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.61 (b, 2H × 2, -OCOCH2CH2(CH2)12CH3), 1.26 (b, 24H × 2, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 2, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); HRMS–FAB (m/z): [M + Na]+calcd for C41H78O10Na, 753.5493;found, 753.5519.
3-O-(6-O-palmitoyl-α-D-glucopyranosyl)-1,2-di-O-palmitoyl-sn-glycerol (11, entry 4): A mixture of 8a (120 mg, 0.12 mmol) and palmitoyl chloride (165 mg, 0.6 mmol) in pyridine was stirred at rt for 3 h and then treated with CH3OH (1 mL) for 3 h. After concentration in vacuo, the residue was purified on silica gel (toluene/ethyl acetate). The main product (138 mg) was dissolved in a mixture of cyclohexene/ethanol 1:4 and subjected to catalytic hydrogenation at atmospheric pressure in the presence of Pd(OH)2/C (50 mg). The product was purified by silica gel column chromatography (CH3OH/CHCl3) to afford 11 (99 mg, 85% yield from 8a). [α]D30 +20.9 (c 1.0, CHCl3/CH3OH 10:1); IR (KBr, film): 3414, 2919, 2851, 1739, 1605, 1465, 1375, 1176, 1054, 720 cm−1; 1H NMR (500 MHz, CDCl3/CD3OD 10:1): δH 5.25 (m, 1H, glycerol H-2), 4.82 (d, 1H, J = 4.0 Hz, H-1), 4.40 (dd, 1H, J = 3.5 and 12.0 Hz, glycerol H-1proS), 4.34 and 4.30 (dd × 2, 2H, J = 5.0 and 12.0, 2.5 and 12.0 Hz, H-6proR and H-6proS), 4.16 (dd, 1H, J = 6.5 and 12.0 Hz, glycerol H-1proR), 3.82 (dd, 1H, J = 5.0 and 10.5 Hz, glycerol H-3proR), 3.73 (m, 1H, H-5), 3.65 (dd, 1H, J = 9.0 and 9.5 Hz, H-3), 3.61 (dd, 1H, J = 5.5 and 10.5 Hz, glycerol H-3proS), 3.45 (dd, 1H, J = 4.0 and 9.5 Hz, H-2), 3.33 (dd, 1H, J = 9.0 and 10.0 Hz, H-4), 2.33 (m, 2H × 3, -OCOCH2CH2(CH2)12CH3), 1.61 (b, 2H × 3, -OCOCH2CH2(CH2)12CH3), 1.26 (b, 24H × 3, -OCOCH2CH2(CH2)12CH3), 0.88 (t, 3H × 3, J = 7.0 Hz, -OCOCH2CH2(CH2)12CH3); HRMS–FAB (m/z): [M + Na]+ calcd for C57H108O11Na, 991.7789; found, 991.7832.
Acknowledgements
This work was supported by the Industrial Technology Research Grant Program from New Energy and Industrial Technology Development Organization (NEDO) of Japan and from the Science & Technology Project for a Safe & Secure Society from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
References
-
Rawadi, G. Microbes Infect. 2000, 2, 955–964. doi:10.1016/S1286-4579(00)00395-6
Return to citation in text: [1] -
Shimizu, T.; Kida, Y.; Kuwano, K. Immunology 2004, 113, 121–129. doi:10.1111/j.1365-2567.2004.01937.x
Return to citation in text: [1] -
Romero, F.; Moreno, E.; Ruiz-Bravo, A.; Jiménez-Valera, M. Curr. Microbiol. 2004, 48, 237–239. doi:10.1007/s00284-003-4134-1
Return to citation in text: [1] -
Rottem, S. Biochim. Biophys. Acta, Rev. Biomembr. 1980, 604, 65–90. doi:10.1016/0005-2736(80)90585-4
Return to citation in text: [1] -
Boggs, J. M. Biochim. Biophys. Acta 1987, 906, 353–404.
Return to citation in text: [1] -
Toujima, S.; Kuwano, K.; Zhang, Y.; Fujimoto, N.; Hirama, M.; Oishi, T.; Fukuda, S.; Nagumo, Y.; Imai, H.; Kikuchi, T.; Arai, S. Microbiology (Reading, U. K.) 2000, 146, 2317–2323.
Return to citation in text: [1] -
Matsuda, K.; Kasama, T.; Ishizuka, I.; Handa, S.; Yamamoto, N.; Taki, T. J. Biol. Chem. 1994, 269, 33123–33128.
Return to citation in text: [1] -
Matsuda, K.; Li, J.-L.; Harasawa, R.; Yamamoto, N. Biochem. Biophys. Res. Commun. 1997, 233, 644–649. doi:10.1006/bbrc.1997.6443
Return to citation in text: [1] -
Li, J.-L.; Matsuda, K.; Takagi, M.; Yamamoto, N. J. Immunol. Methods 1997, 208, 103–113. doi:10.1016/S0022-1759(97)00135-X,
Return to citation in text: [1] -
Matsuda, K.; Li, J.-L.; Ichinose, S.; Harasawa, R.; Saito, M.; Yamamoto, N. Microbiol. Immunol. 2000, 44, 695–702.
Return to citation in text: [1] -
Zähringer, U.; Wagner, F.; Rietschel, E. T.; Ben-Menachem, G.; Deutsch, J.; Rottem, S. J. Biol. Chem. 1997, 272, 26262–26270. doi:10.1074/jbc.272.42.26262
Return to citation in text: [1] -
Ben-Menachem, G.; Byström, T.; Rechnitzer, H.; Rottem, S.; Rilfors, L.; Lindblom, G. Eur. J. Biochem. 2001, 268, 3694–3701. doi:10.1046/j.1432-1327.2001.02277.x
Return to citation in text: [1] [2] -
Brandenburg, K.; Wagner, F.; Müller, M.; Heine, H.; Andrä, J.; Koch, M. H. J.; Zähringer, U.; Seydel, U. Eur. J. Biochem. 2003, 270, 3271–3279. doi:10.1046/j.1432-1033.2003.03719.x
Return to citation in text: [1] [2] -
Yavlovich, A.; Katzenell, A.; Tarshis, M.; Higazi, A. A.-R.; Rottem, S. Infect. Immun. 2004, 72, 5004–5011. doi:10.1128/IAI.72.9.5004-5011.2004
Return to citation in text: [1] -
Nishida, Y.; Ohrui, H.; Meguro, H.; Ishizawa, M.; Matsuda, K.; Taki, T.; Handa, S.; Yamamoto, N. Tetrahedron Lett. 1994, 35, 5465–5468. doi:10.1016/S0040-4039(00)73526-X
Return to citation in text: [1] [2] -
Nishida, Y.; Takamori, Y.; Ohrui, H.; Ishizaka, I.; Matsuda, K.; Kobayashi, K. Tetrahedron Lett. 1999, 40, 2371–2374. doi:10.1016/S0040-4039(99)00191-4
Return to citation in text: [1] [2] -
Matsuda, K. Phosphocholine-containing glycoglycerolipids of Mycoplasma fermentans as a pathogen of rheumatoid arthritis: possible role of Mycoplasma fermentans GGPLs in the pathogenesis of neuroendocrine-immune abnormalities.. In Recent research developments in neuroscience; Pandalai, S. G., Ed.; Research Signpost: India, 2004; Vol. 1, pp 15–23.
Return to citation in text: [1] -
Kawahito, Y.; Ichinose, S.; Sano, H.; Tsubouchi, Y.; Kohno, M.; Yoshikawa, T.; Tokunaga, D.; Hojo, T.; Harasawa, R.; Nakano, T.; Matsuda, K. Biochem. Biophys. Res. Commun. 2008, 369, 561–566. doi:10.1016/j.bbrc.2008.02.079
Return to citation in text: [1] -
Sato, N.; Oizumi, T.; Kinbara, M.; Sato, T.; Funayama, H.; Sato, S.; Matsuda, K.; Takada, H.; Sugawara, S.; Endo, Y. FEMS Immunol. Med. Microbiol. 2010, 59, 33–41. doi:10.1111/j.1574-695X.2010.00657.x
Return to citation in text: [1] -
Fujiwara, M.; Ishida, N.; Asano, K.; Matsuda, K.; Nomura, N.; Nishida, Y.; Harasawa, R. J. Vet. Med. Sci. 2010, 72, 805–808. doi:10.1292/jvms.09-0572
Return to citation in text: [1] -
Ishida, N.; Irikura, D.; Matsuda, K.; Sato, S.; Sone, T.; Tanaka, M.; Asano, K. Curr. Microbiol. 2009, 58, 535–540. doi:10.1007/s00284-009-9362-6
Return to citation in text: [1] -
Ishida, N.; Irikura, D.; Matsuda, K.; Sato, S.; Sone, T.; Tanaka, M.; Asano, K. J. Biosci. Bioeng. 2010, 109, 341–345. doi:10.1016/j.jbiosc.2009.09.049
Return to citation in text: [1] -
Shingu, Y.; Nishida, Y.; Dohi, H.; Matsuda, K.; Kobayashi, K. J. Carbohydr. Chem. 2002, 21, 605–611. doi:10.1081/CAR-120016858
Return to citation in text: [1] [2] [3] -
Shingu, Y.; Nishida, Y.; Dohi, H.; Kobayashi, K. Org. Biomol. Chem. 2003, 1, 2518–2521. doi:10.1039/b303984f
Return to citation in text: [1] -
Nishida, Y.; Shingu, Y.; Dohi, H.; Kobayashi, K. Org. Lett. 2003, 5, 2377–2380. doi:10.1021/ol034269+
Return to citation in text: [1] [2] -
Shingu, Y.; Miyachi, A.; Miura, Y.; Kobayashi, K.; Nishida, Y. Carbohydr. Res. 2005, 340, 2236–2244. doi:10.1016/j.carres.2005.07.020
Return to citation in text: [1] [2] -
Gigg, R.; Penglis, A. A. E.; Conant, R. J. Chem. Soc., Perkin Trans. 1 1977, 2014–2017. doi:10.1039/P19770002014
Return to citation in text: [1] -
Mannock, D. A.; Lewis, R. N.; McElhaney, R. N. Chem. Phys. Lipids 1990, 55, 309–321. doi:10.1016/0009-3084(90)90169-R
Return to citation in text: [1] -
Gordon, D. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1992, 114, 659–663. doi:10.1021/ja00028a036
Return to citation in text: [1] [2] [3] -
Imai, H.; Oishi, T.; Kikuchi, T.; Hirama, M. Tetrahedron 2000, 56, 8451–8459. doi:10.1016/S0040-4020(00)00793-6
Return to citation in text: [1] [2] [3] -
Shvets, V. I.; Bashkatova, A. I.; Evstigneeva, R. P. Chem. Phys. Lipids 1973, 10, 267–285. doi:10.1016/0009-3084(73)90006-6
And see the related references cited therein.
Return to citation in text: [1] -
Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032
Return to citation in text: [1] -
Lu, S.-R.; Lay, Y.-H.; Chen, J.-H.; Liu, C.-Y.; Mong, K.-K. T. Angew. Chem. 2011, 123, 7453–7458. doi:10.1002/ange.201100076
And see the related references cited therein.
Return to citation in text: [1] -
Hayakawa, Y.; Kataoka, M. J. Am. Chem. Soc. 1997, 119, 11758–11762. doi:10.1021/ja970685b
Return to citation in text: [1] -
Nishida, Y.; Uzawa, H.; Hanada, S.; Ohrui, H.; Meguro, H. Agric. Biol. Chem. 1989, 53, 2319–2326. doi:10.1271/bbb1961.53.2319
Return to citation in text: [1] -
Uzawa, H.; Nishida, Y.; Hanada, S.; Ohrui, H.; Meguro, H. J. Chem. Soc., Chem. Commun. 1989, 862–863. doi:10.1039/C39890000862
Return to citation in text: [1] -
Uzawa, H.; Nishida, Y.; Ohrui, H.; Meguro, H. J. Org. Chem. 1990, 55, 116–122. doi:10.1021/jo00288a024
Return to citation in text: [1] -
Ohrui, H.; Nishida, Y.; Watanabe, M.; Hori, H.; Meguro, H. Tetrahedron Lett. 1985, 26, 3251–3254. doi:10.1016/S0040-4039(00)98164-4
Return to citation in text: [1] -
Nishida, Y.; Hori, H.; Ohrui, H.; Meguro, H.; Uzawa, J.; Reimer, D.; Sinnwell, V.; Paulsen, H. Tetrahedron Lett. 1988, 29, 4461–4464. doi:10.1016/S0040-4039(00)80522-5
Return to citation in text: [1] -
Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H.; Uzawa, J. Tetrahedron Lett. 1988, 29, 4457–4460. doi:10.1016/S0040-4039(00)80521-3
Return to citation in text: [1] -
Nishida, Y.; Hori, H.; Ohrui, H.; Meguro, H. J. Carbohydr. Chem. 1988, 7, 239–250. doi:10.1080/07328308808058917
Return to citation in text: [1] -
Haasnoot, C. A. G.; de Leeuw, F. A. A. M.; Altona, C. Tetrahedron 1980, 36, 2783–2790. doi:10.1016/0040-4020(80)80155-4
Return to citation in text: [1] -
Matsuda, K. Antigens: Lipids. In Encyclopedia of Life Science; John Wiley and Sons: Chichester, U. K., 2002; pp. 748–755.
Return to citation in text: [1] -
Koynova, R. D.; Kuttenreich, H. L.; Tenchov, B. G.; Hinz, H. J. Biochemistry 1988, 27, 4612–4619. doi:10.1021/bi00413a005
Return to citation in text: [1] -
Mannock, D. A.; Lewis, R. N.; McElhaney, R. N.; Akiyama, M.; Yamada, H.; Turner, D. C.; Gruner, S. M. Biophys. J. 1992, 63, 1355–1368. doi:10.1016/S0006-3495(92)81713-7
Return to citation in text: [1]
| 44. | Koynova, R. D.; Kuttenreich, H. L.; Tenchov, B. G.; Hinz, H. J. Biochemistry 1988, 27, 4612–4619. doi:10.1021/bi00413a005 |
| 45. | Mannock, D. A.; Lewis, R. N.; McElhaney, R. N.; Akiyama, M.; Yamada, H.; Turner, D. C.; Gruner, S. M. Biophys. J. 1992, 63, 1355–1368. doi:10.1016/S0006-3495(92)81713-7 |
| 1. | Rawadi, G. Microbes Infect. 2000, 2, 955–964. doi:10.1016/S1286-4579(00)00395-6 |
| 11. | Zähringer, U.; Wagner, F.; Rietschel, E. T.; Ben-Menachem, G.; Deutsch, J.; Rottem, S. J. Biol. Chem. 1997, 272, 26262–26270. doi:10.1074/jbc.272.42.26262 |
| 12. | Ben-Menachem, G.; Byström, T.; Rechnitzer, H.; Rottem, S.; Rilfors, L.; Lindblom, G. Eur. J. Biochem. 2001, 268, 3694–3701. doi:10.1046/j.1432-1327.2001.02277.x |
| 13. | Brandenburg, K.; Wagner, F.; Müller, M.; Heine, H.; Andrä, J.; Koch, M. H. J.; Zähringer, U.; Seydel, U. Eur. J. Biochem. 2003, 270, 3271–3279. doi:10.1046/j.1432-1033.2003.03719.x |
| 14. | Yavlovich, A.; Katzenell, A.; Tarshis, M.; Higazi, A. A.-R.; Rottem, S. Infect. Immun. 2004, 72, 5004–5011. doi:10.1128/IAI.72.9.5004-5011.2004 |
| 29. | Gordon, D. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1992, 114, 659–663. doi:10.1021/ja00028a036 |
| 30. | Imai, H.; Oishi, T.; Kikuchi, T.; Hirama, M. Tetrahedron 2000, 56, 8451–8459. doi:10.1016/S0040-4020(00)00793-6 |
| 7. | Matsuda, K.; Kasama, T.; Ishizuka, I.; Handa, S.; Yamamoto, N.; Taki, T. J. Biol. Chem. 1994, 269, 33123–33128. |
| 8. | Matsuda, K.; Li, J.-L.; Harasawa, R.; Yamamoto, N. Biochem. Biophys. Res. Commun. 1997, 233, 644–649. doi:10.1006/bbrc.1997.6443 |
| 9. | Li, J.-L.; Matsuda, K.; Takagi, M.; Yamamoto, N. J. Immunol. Methods 1997, 208, 103–113. doi:10.1016/S0022-1759(97)00135-X, |
| 10. | Matsuda, K.; Li, J.-L.; Ichinose, S.; Harasawa, R.; Saito, M.; Yamamoto, N. Microbiol. Immunol. 2000, 44, 695–702. |
| 23. | Shingu, Y.; Nishida, Y.; Dohi, H.; Matsuda, K.; Kobayashi, K. J. Carbohydr. Chem. 2002, 21, 605–611. doi:10.1081/CAR-120016858 |
| 29. | Gordon, D. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1992, 114, 659–663. doi:10.1021/ja00028a036 |
| 30. | Imai, H.; Oishi, T.; Kikuchi, T.; Hirama, M. Tetrahedron 2000, 56, 8451–8459. doi:10.1016/S0040-4020(00)00793-6 |
| 31. |
Shvets, V. I.; Bashkatova, A. I.; Evstigneeva, R. P. Chem. Phys. Lipids 1973, 10, 267–285. doi:10.1016/0009-3084(73)90006-6
And see the related references cited therein. |
| 4. | Rottem, S. Biochim. Biophys. Acta, Rev. Biomembr. 1980, 604, 65–90. doi:10.1016/0005-2736(80)90585-4 |
| 5. | Boggs, J. M. Biochim. Biophys. Acta 1987, 906, 353–404. |
| 6. | Toujima, S.; Kuwano, K.; Zhang, Y.; Fujimoto, N.; Hirama, M.; Oishi, T.; Fukuda, S.; Nagumo, Y.; Imai, H.; Kikuchi, T.; Arai, S. Microbiology (Reading, U. K.) 2000, 146, 2317–2323. |
| 25. | Nishida, Y.; Shingu, Y.; Dohi, H.; Kobayashi, K. Org. Lett. 2003, 5, 2377–2380. doi:10.1021/ol034269+ |
| 26. | Shingu, Y.; Miyachi, A.; Miura, Y.; Kobayashi, K.; Nishida, Y. Carbohydr. Res. 2005, 340, 2236–2244. doi:10.1016/j.carres.2005.07.020 |
| 2. | Shimizu, T.; Kida, Y.; Kuwano, K. Immunology 2004, 113, 121–129. doi:10.1111/j.1365-2567.2004.01937.x |
| 3. | Romero, F.; Moreno, E.; Ruiz-Bravo, A.; Jiménez-Valera, M. Curr. Microbiol. 2004, 48, 237–239. doi:10.1007/s00284-003-4134-1 |
| 27. | Gigg, R.; Penglis, A. A. E.; Conant, R. J. Chem. Soc., Perkin Trans. 1 1977, 2014–2017. doi:10.1039/P19770002014 |
| 28. | Mannock, D. A.; Lewis, R. N.; McElhaney, R. N. Chem. Phys. Lipids 1990, 55, 309–321. doi:10.1016/0009-3084(90)90169-R |
| 29. | Gordon, D. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1992, 114, 659–663. doi:10.1021/ja00028a036 |
| 30. | Imai, H.; Oishi, T.; Kikuchi, T.; Hirama, M. Tetrahedron 2000, 56, 8451–8459. doi:10.1016/S0040-4020(00)00793-6 |
| 17. | Matsuda, K. Phosphocholine-containing glycoglycerolipids of Mycoplasma fermentans as a pathogen of rheumatoid arthritis: possible role of Mycoplasma fermentans GGPLs in the pathogenesis of neuroendocrine-immune abnormalities.. In Recent research developments in neuroscience; Pandalai, S. G., Ed.; Research Signpost: India, 2004; Vol. 1, pp 15–23. |
| 20. | Fujiwara, M.; Ishida, N.; Asano, K.; Matsuda, K.; Nomura, N.; Nishida, Y.; Harasawa, R. J. Vet. Med. Sci. 2010, 72, 805–808. doi:10.1292/jvms.09-0572 |
| 21. | Ishida, N.; Irikura, D.; Matsuda, K.; Sato, S.; Sone, T.; Tanaka, M.; Asano, K. Curr. Microbiol. 2009, 58, 535–540. doi:10.1007/s00284-009-9362-6 |
| 22. | Ishida, N.; Irikura, D.; Matsuda, K.; Sato, S.; Sone, T.; Tanaka, M.; Asano, K. J. Biosci. Bioeng. 2010, 109, 341–345. doi:10.1016/j.jbiosc.2009.09.049 |
| 12. | Ben-Menachem, G.; Byström, T.; Rechnitzer, H.; Rottem, S.; Rilfors, L.; Lindblom, G. Eur. J. Biochem. 2001, 268, 3694–3701. doi:10.1046/j.1432-1327.2001.02277.x |
| 13. | Brandenburg, K.; Wagner, F.; Müller, M.; Heine, H.; Andrä, J.; Koch, M. H. J.; Zähringer, U.; Seydel, U. Eur. J. Biochem. 2003, 270, 3271–3279. doi:10.1046/j.1432-1033.2003.03719.x |
| 23. | Shingu, Y.; Nishida, Y.; Dohi, H.; Matsuda, K.; Kobayashi, K. J. Carbohydr. Chem. 2002, 21, 605–611. doi:10.1081/CAR-120016858 |
| 24. | Shingu, Y.; Nishida, Y.; Dohi, H.; Kobayashi, K. Org. Biomol. Chem. 2003, 1, 2518–2521. doi:10.1039/b303984f |
| 16. | Nishida, Y.; Takamori, Y.; Ohrui, H.; Ishizaka, I.; Matsuda, K.; Kobayashi, K. Tetrahedron Lett. 1999, 40, 2371–2374. doi:10.1016/S0040-4039(99)00191-4 |
| 15. | Nishida, Y.; Ohrui, H.; Meguro, H.; Ishizawa, M.; Matsuda, K.; Taki, T.; Handa, S.; Yamamoto, N. Tetrahedron Lett. 1994, 35, 5465–5468. doi:10.1016/S0040-4039(00)73526-X |
| 18. | Kawahito, Y.; Ichinose, S.; Sano, H.; Tsubouchi, Y.; Kohno, M.; Yoshikawa, T.; Tokunaga, D.; Hojo, T.; Harasawa, R.; Nakano, T.; Matsuda, K. Biochem. Biophys. Res. Commun. 2008, 369, 561–566. doi:10.1016/j.bbrc.2008.02.079 |
| 19. | Sato, N.; Oizumi, T.; Kinbara, M.; Sato, T.; Funayama, H.; Sato, S.; Matsuda, K.; Takada, H.; Sugawara, S.; Endo, Y. FEMS Immunol. Med. Microbiol. 2010, 59, 33–41. doi:10.1111/j.1574-695X.2010.00657.x |
| 33. |
Lu, S.-R.; Lay, Y.-H.; Chen, J.-H.; Liu, C.-Y.; Mong, K.-K. T. Angew. Chem. 2011, 123, 7453–7458. doi:10.1002/ange.201100076
And see the related references cited therein. |
| 23. | Shingu, Y.; Nishida, Y.; Dohi, H.; Matsuda, K.; Kobayashi, K. J. Carbohydr. Chem. 2002, 21, 605–611. doi:10.1081/CAR-120016858 |
| 32. | Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. doi:10.1021/ja00847a032 |
| 42. | Haasnoot, C. A. G.; de Leeuw, F. A. A. M.; Altona, C. Tetrahedron 1980, 36, 2783–2790. doi:10.1016/0040-4020(80)80155-4 |
| 43. | Matsuda, K. Antigens: Lipids. In Encyclopedia of Life Science; John Wiley and Sons: Chichester, U. K., 2002; pp. 748–755. |
| 38. | Ohrui, H.; Nishida, Y.; Watanabe, M.; Hori, H.; Meguro, H. Tetrahedron Lett. 1985, 26, 3251–3254. doi:10.1016/S0040-4039(00)98164-4 |
| 39. | Nishida, Y.; Hori, H.; Ohrui, H.; Meguro, H.; Uzawa, J.; Reimer, D.; Sinnwell, V.; Paulsen, H. Tetrahedron Lett. 1988, 29, 4461–4464. doi:10.1016/S0040-4039(00)80522-5 |
| 40. | Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H.; Uzawa, J. Tetrahedron Lett. 1988, 29, 4457–4460. doi:10.1016/S0040-4039(00)80521-3 |
| 41. | Nishida, Y.; Hori, H.; Ohrui, H.; Meguro, H. J. Carbohydr. Chem. 1988, 7, 239–250. doi:10.1080/07328308808058917 |
| 15. | Nishida, Y.; Ohrui, H.; Meguro, H.; Ishizawa, M.; Matsuda, K.; Taki, T.; Handa, S.; Yamamoto, N. Tetrahedron Lett. 1994, 35, 5465–5468. doi:10.1016/S0040-4039(00)73526-X |
| 16. | Nishida, Y.; Takamori, Y.; Ohrui, H.; Ishizaka, I.; Matsuda, K.; Kobayashi, K. Tetrahedron Lett. 1999, 40, 2371–2374. doi:10.1016/S0040-4039(99)00191-4 |
| 35. | Nishida, Y.; Uzawa, H.; Hanada, S.; Ohrui, H.; Meguro, H. Agric. Biol. Chem. 1989, 53, 2319–2326. doi:10.1271/bbb1961.53.2319 |
| 36. | Uzawa, H.; Nishida, Y.; Hanada, S.; Ohrui, H.; Meguro, H. J. Chem. Soc., Chem. Commun. 1989, 862–863. doi:10.1039/C39890000862 |
| 37. | Uzawa, H.; Nishida, Y.; Ohrui, H.; Meguro, H. J. Org. Chem. 1990, 55, 116–122. doi:10.1021/jo00288a024 |
| 25. | Nishida, Y.; Shingu, Y.; Dohi, H.; Kobayashi, K. Org. Lett. 2003, 5, 2377–2380. doi:10.1021/ol034269+ |
| 26. | Shingu, Y.; Miyachi, A.; Miura, Y.; Kobayashi, K.; Nishida, Y. Carbohydr. Res. 2005, 340, 2236–2244. doi:10.1016/j.carres.2005.07.020 |
| 34. | Hayakawa, Y.; Kataoka, M. J. Am. Chem. Soc. 1997, 119, 11758–11762. doi:10.1021/ja970685b |
© 2012 Nishida et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)