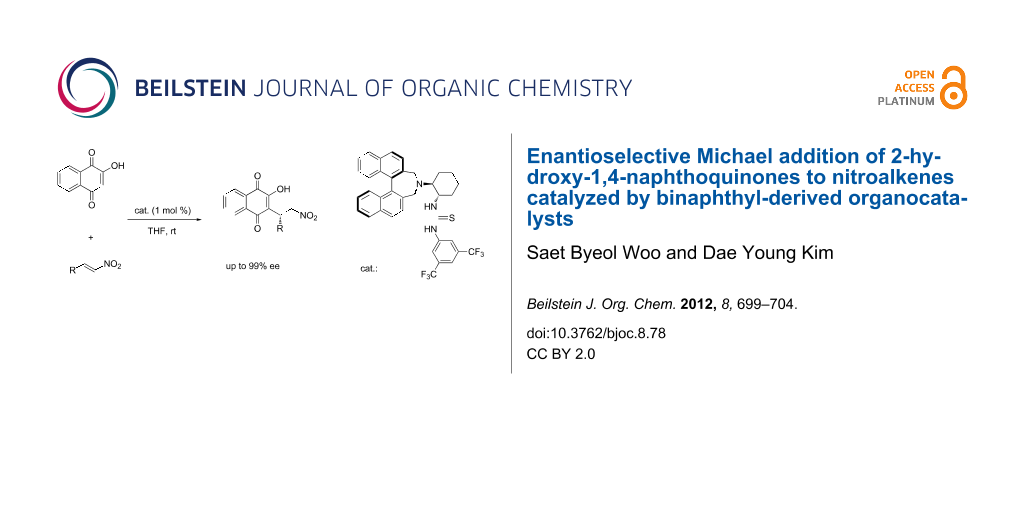
Abstract
The highly enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinones to nitroalkenes, promoted by binaphthyl-modified chiral bifunctional organocatalysts is described. This reaction afforded the chiral functionalized naphthoquinones in high yields (81–95%) and excellent enantioselectivities (91–98% ee) under low catalyst loading (1 mol %).

Graphical Abstract
Introduction
Quinone and naphthoquinone structures exist in a large number of natural products and biologically active molecules [1-4]. Many of these naturally occurring naphthoquinones and their synthetic analogues are important precursors for the synthesis of natural products and pharmaceuticals [5-9]. The stereoselective formation of C–C bonds is of great importance for the synthesis of enantiomerically pure, biologically active organic compounds [10,11]. It is widely recognized that the Michael addition is one of the most versatile and general methods for C–C bond formation in organic synthesis [12], and intensive research efforts have been directed toward the development of enantioselective catalytic protocols for this reaction [13-15]. The organocatalyst-mediated enantioselective conjugate addition reactions, which are both powerful and environmentally friendly, have been subjected to rigorous investigation in recent years [16-22]. The asymmetric Michael addition of various nucleophiles to nitroalkenes is of great interest, because the products obtained are versatile intermediates in organic synthesis [23-26]. Extensive studies have been devoted to the development of asymmetric conjugate additions of 1,3-dicarbonyl compounds to various Michael acceptors [27-33]. Recently, the groups of Du and Zhou reported a highly enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinones to nitroalkenes catalyzed by chiral, bifunctional tertiary-amine thioureas, thiophosphorodiamides, and squaramide-based organocatalysts [34-36].
Findings
In the framework of our research program for the development of synthetic methods for the enantioselective construction of stereogenic carbon centers [37-42], we recently reported the enantioselective Michael addition of active methines to nitroalkenes [43,44]. Herein, we describe the direct enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinone with nitroalkenes, catalyzed by bifunctional organocatalysts (Figure 1) that bear both central and axial chiral elements [45-47].
![[1860-5397-8-78-1]](/bjoc/content/figures/1860-5397-8-78-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of chiral organocatalysts.
Figure 1: Structures of chiral organocatalysts.
We initially investigated the reaction system with 2-hydroxy-1,4-naphthoquinone (1) and nitrostyrene 2a in the presence of 10 mol % of Takemoto's catalyst I in acetonitrile at room temperature, to determine the optimum reaction conditions for the catalytic, enantioselective Michael addition. This reaction exhibited good yield and high enantioselectivity (89% ee, Table 1, entry 1). In order to enhance the enantioselectivity, other bifunctional organocatalysts II–VIII were evaluated in the model reaction (Table 1, entries 2–8). The quinine-derived thiourea catalyst II was less effective (Table 1, entries 1 and 2), whereas the binaphthyl-modified, chiral, bifunctional organocatalysts III–VIII, bearing both central and axial chiral elements, effectively promoted the addition reaction in high yield, with high enantioselectivity (78–97% ee, Table 1, entries 3–8). Catalyst III gave the desired product 3a with high enantioselectivity (97%, Table 1, entry 3), whereas the diastereomeric catalyst VII afforded product 3a in lower enantioselectivity (78% ee, Table 1, entry 7). These results demonstrate that the central and axial chiral elements in the chiral amine-thiourea catalyst III are matched, thus enhancing the stereochemical control, whereas in the diastereomeric catalyst VII this is not the case.
Table 1: Optimization of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-8-78-i1.svg?max-width=637&scale=1.0)
|
|||||
| entry | cat. | solvent | time (h) | yield (%)a | ee (%)b |
|---|---|---|---|---|---|
| 1 | I | CH3CN | 2 | 84 | 89 |
| 2 | II | CH3CN | 2 | 87 | 77 |
| 3 | III | CH3CN | 2 | 96 | 97 |
| 4 | IV | CH3CN | 2 | 95 | 87 |
| 5 | V | CH3CN | 2 | 93 | 81 |
| 6 | VI | CH3CN | 2 | 90 | 93 |
| 7 | VII | CH3CN | 2 | 85 | 78 |
| 8 | VIII | CH3CN | 2 | 88 | 93 |
| 9 | III | toluene | 4 | 75 | 95 |
| 10 | III | DCM | 4 | 93 | 89 |
| 11 | III | THF | 2 | 92 | 99 |
| 12 | III | Et2O | 3 | 81 | 91 |
| 13 | III | H2O | 17 | 89 | 19 |
| 14 | III | brine | 17 | 86 | 37 |
| 15c | III | THF | 2 | 90 | 98 |
| 16d | III | THF | 2 | 90 | 99 |
| 17e | III | THF | 2 | 89 | 99 |
aIsolated yield.
bEnantiopurity was determined by HPLC analysis using chiralcel OJ-H column.
cReaction was carried out in the presence of 5 mol % catalyst.
dReaction was carried out in the presence of 2.5 mol % catalyst.
eReaction was carried out in the presence of 1 mol % catalyst.
Different solvents were then tested in the presence of 10 mol % of catalyst III together with 2-hydroxy-1,4-naphthoquinone (1) and nitrostyrene 2a in order to further improve the selectivity of the reaction. Aprotic solvents, such as acetonitrile, toluene, dichloromethane, THF, diethyl ether, were well tolerated in this conjugate addition without a significant decrease of enantioselectivities (89–99% ee, Table 1, entries 3 and 9–12). Remarkably, water and brine also afforded products in good yields; however, the selectivity dropped significantly (Table 1, entries 13 and 14). Among the solvents probed, the best results (92% yield and 99% ee) were achieved when the reaction was conducted in THF (Table 1, entry 11). The present catalytic system tolerates catalyst loading down to 5, 2.5, and 1 mol % without compromising the yield or enantioselectivity (Table 1, entries 11 and 15–17).
With the optimized reaction conditions in hand, the scope of the methodology was investigated in reactions with 2-hydroxy-1,4-naphthoquinone (1) and various nitroalkenes 2a–l in the presence of 1 mol % of catalyst III in THF at room temperature (Table 2). A range of electron-donating and electron-withdrawing substitutions on the β-aryl ring of the nitrostyrenes 2b–h provided reaction products in high yields and excellent enantioselectivities. Heteroaryl- and naphthyl-substituted nitroalkenes 2i and 2j provided products with high selectivity (93–99% ee, Table 2, entries 9 and 10). The β-alkyl-substituted nitroalkene, 4-methyl-1-nitropent-1-ene (2k), was also an acceptable starting material and provided the corresponding Michael adducts in high yield and excellent enantioeselectivity (97% ee, Table 2, entry 11).
Table 2: Catalytic asymmetric Michael addition of 2-hydroxy-1,4-naphthoquinone 1 to nitroalkenes 2.
![[Graphic 2]](/bjoc/content/inline/1860-5397-8-78-i2.svg?max-width=637&scale=1.0)
|
||||
| entry | 2, R | time (h) | yield (%)a | ee (%)b |
|---|---|---|---|---|
| 1 | 2a, Ph | 2 | 3a, 89 | 99 |
| 2 | 2b, p-MeC6H4 | 2 | 3b, 93 | 95 |
| 3 | 2c, p-MeOC6H4 | 4 | 3c, 81 | 99 |
| 4 | 2d, p-FC6H4 | 3 | 3d, 95 | 95 |
| 5 | 2e, p-ClC6H4 | 3 | 3e, 90 | 91 |
| 6 | 2f, p-BrC6H4 | 3 | 3f, 95 | 95 |
| 7 | 2g, o-FC6H4 | 4 | 3g, 95 | 95 |
| 8 | 2h, o-BrC6H4 | 4 | 3h, 95 | 95 |
| 9 | 2i, 2-thienyl | 5 | 3i, 93 | 93 |
| 10 | 2j, 2-naphthyl | 5 | 3j, 93 | 99 |
| 11 | 2k, isobutyl | 5 | 3k, 90 | 97 |
aIsolated yield.
bEnantiopurity was determined by HPLC analysis using chiralcel OJ-H (3a–j) and chiralpak AD-H (for 3k) columns.
In conclusion, we have developed a highly efficient catalytic, enantioselective Michael addition of 2-hydroxy-1,4-naphthoquinone to nitroalkenes using a binaphthyl-derived tertiary amine-thiourea organocatalyst. The various types of nitroalkylated naphthoquinone derivatives were obtained in good to high yields with excellent enantioselectivities (91–99% ee) for all the substrates examined in this work. We believe that this method should provide a practical entry for the preparation of chiral nitroalkylated naphthoquinone derivatives. Further details and application of this asymmetric Michael addition of 2-hydroxy-1,4-naphthoquinone nucleophiles will be presented in due course.
Experimental
General procedure for the Michael addition of 2-hydroxy-1,4-naphthoquinone (1) with nitroalkenes 2: A mixture of 2-hydroxy-1,4-naphthoquinones (1, 34.8 mg, 0.2 mmol) and catalyst III (1.3 mg, 0.002 mmol) in THF (0.4 mL) was stirred at room temperature for 5 min. A solution of nitroalkene 2 (0.2 mmol) was added. The reaction mixture was stirred for 2–5 h at room temperature. After completion of the reaction, the resulting solution was concentrated in vacuo and the obtained residue was purified by flash chromatography (EtOAc–hexane) to afford the corresponding Michael adducts 3. Products 3 are known compounds, and their data were identical to those reported in the literature [34-36].
Supporting Information
| Supporting Information File 1: Characterization data of products 3. | ||
| Format: PDF | Size: 206.2 KB | Download |
References
-
de Andrade-Neto, V. F.; Goulart, M. O. F.; da Silva Filho, J. F.; da Silva, M. J.; Pinto, M. C. F. R.; Pinto, A. V.; Zalis, M. G.; Carvalho, L. H.; Krettli, A. U. Bioorg. Med. Chem. Lett. 2004, 14, 1145–1149. doi:10.1016/j.bmcl.2003.12.069
Return to citation in text: [1] -
Tandon, V. K.; Yadav, D. B.; Singh, R. V.; Chaturvedi, A. K.; Shukla, P. K. Bioorg. Med. Chem. Lett. 2005, 15, 5324–5328. doi:10.1016/j.bmcl.2005.08.032
Return to citation in text: [1] -
Glänzel, M.; Bültmann, R.; Starke, K.; Frahm, A. W. Eur. J. Med. Chem. 2005, 40, 1262–1276. doi:10.1016/j.ejmech.2005.07.007
Return to citation in text: [1] -
Gomez-Monterrey, I.; Santelli, G.; Campiglia, P.; Califano, D.; Falasconi, F.; Pisano, C.; Vesci, L.; Lama, T.; Grieco, P.; Novellino, E. J. Med. Chem. 2005, 48, 1152–1157. doi:10.1021/jm0408565
Return to citation in text: [1] -
Gomez-Monterrey, I.; Campiglia, P.; Carotenuto, A.; Califano, D.; Pisano, C.; Vesci, L.; Lama, T.; Bertamino, A.; Sala, M.; Mazzella di Bosco, A.; Grieco, P.; Novellino, E. J. Med. Chem. 2007, 50, 1787–1798. doi:10.1021/jm0612158
Return to citation in text: [1] -
Castellano, S.; Bertamino, A.; Gomez-Monterrey, I.; Santoriello, M.; Grieco, P.; Campiglia, P.; Sbardella, G.; Novellino, E. Tetrahedron Lett. 2008, 49, 583–585. doi:10.1016/j.tetlet.2007.11.148
Return to citation in text: [1] -
Hsin, L.-W.; Wang, H.-P.; Kao, P.-H.; Lee, O.; Chen, W.-R.; Chen, H.-W.; Guh, J.-H.; Chan, Y.-L.; His, C.-P.; Yang, M.-S.; Li, T.-K.; Lee, C.-H. Bioorg. Med. Chem. 2008, 16, 1006–1014. doi:10.1016/j.bmc.2007.10.012
Return to citation in text: [1] -
Wei, P.; Zhang, X.; Tu, S.; Yan, S.; Ying, H.; Ouyang, P. Bioorg. Med. Chem. Lett. 2009, 19, 828–830. doi:10.1016/j.bmcl.2008.12.006
Return to citation in text: [1] -
Zhang, G.; Wang, Y.; Zhang, W.; Xu, X.; Zhong, A.; Xu, D. Eur. J. Org. Chem. 2011, 2142–2147. doi:10.1002/ejoc.201001570
Return to citation in text: [1] -
Corey, E. J.; Guzman-Perez, A. Angew. Chem., Int. Ed. 1998, 37, 388–401. doi:10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V
Return to citation in text: [1] -
Christoffers, J.; Mann, A. Angew. Chem., Int. Ed. 2001, 40, 4591–4597. doi:10.1002/1521-3773(20011217)40:24<4591::AID-ANIE4591>3.0.CO;2-V
Return to citation in text: [1] -
Leonard, J. Contemp. Org. Synth. 1994, 1, 387–415. doi:10.1039/CO9940100387
Return to citation in text: [1] -
Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171–196. doi:10.1055/s-2001-10803
Return to citation in text: [1] -
Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877–1894. doi:10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U
Return to citation in text: [1] -
Christoffers, J.; Baro, A. Angew. Chem., Int. Ed. 2003, 42, 1688–1690. doi:10.1002/anie.200201614
Return to citation in text: [1] -
Connon, S. J. Angew. Chem., Int. Ed. 2006, 45, 3909–3912. doi:10.1002/anie.200600529
Return to citation in text: [1] -
Tylor, M. S.; Jacobson, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] -
Yu, X.; Wang, W. Chem.–Asian J. 2008, 3, 516–532. doi:10.1002/asia.200700415
Return to citation in text: [1] -
Connon, S. J. Synlett 2009, 354–376. doi:10.1055/s-0028-1087557
Return to citation in text: [1] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Nájera, D. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] -
Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001.
Return to citation in text: [1] -
Calderari, G.; Seebach, D. Helv. Chim. Acta 1985, 68, 1592–1604. doi:10.1002/hlca.19850680611
Return to citation in text: [1] -
Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017–1047. doi:10.1016/j.tet.2003.11.016
Return to citation in text: [1] -
Czekelius, C.; Carreira, E. M. Angew. Chem., Int. Ed. 2005, 44, 612–615. doi:10.1002/anie.200461879
Return to citation in text: [1] -
Hamashima, Y.; Hotta, D.; Sodeoka, M. J. Am. Chem. Soc. 2002, 124, 11240–11241. doi:10.1021/ja027075i
Return to citation in text: [1] -
Wu, F.; Li, H.; Hong, R.; Deng, L. Angew. Chem., Int. Ed. 2006, 45, 947–950. doi:10.1002/anie.200502658
Return to citation in text: [1] -
Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2006, 47, 4565–4568. doi:10.1016/j.tetlet.2006.05.003
Return to citation in text: [1] -
Bartoli, G.; Bosco, M.; Carlone, A.; Cavalli, A.; Locatelli, M.; Mazzanti, A.; Ricci, P.; Sambri, L.; Melchiorre, P. Angew. Chem., Int. Ed. 2006, 45, 4966–4970. doi:10.1002/anie.200600370
Return to citation in text: [1] -
Rigby, C. L.; Dixon, D. J. Chem. Commun. 2008, 3798–3800. doi:10.1039/B805233F
Return to citation in text: [1] -
Jung, S. H.; Kim, D. Y. Tetrahedron Lett. 2008, 49, 5527–5530. doi:10.1016/j.tetlet.2008.07.041
Return to citation in text: [1] -
Capuzzi, M.; Perdicchia, D.; Jørgensen, K. A. Chem.–Eur. J. 2008, 14, 128–135. doi:10.1002/chem.200701317
Return to citation in text: [1] -
Zhou, W.-M.; Liu, H.; Du, D.-M. Org. Lett. 2008, 10, 2817–2820. doi:10.1021/ol800945e
Return to citation in text: [1] [2] -
Wu, R.; Chang, X.; Lu, A.; Wang, Y.; Wu, G.; Song, H.; Zhou, Z.; Tang, C. Chem. Commun. 2011, 47, 5034–5036. doi:10.1039/c1cc10797f
Return to citation in text: [1] [2] -
Yang, W.; Du, D.-M. Adv. Synth. Catal. 2011, 353, 1241–1246. doi:10.1002/adsc.201000981
Return to citation in text: [1] [2] -
Kang, Y. K.; Kwon, B. K.; Mang, J. Y.; Kim, D. Y. Tetrahedron Lett. 2011, 52, 3247–3249. doi:10.1016/j.tetlet.2011.04.084
Return to citation in text: [1] -
Kang, Y. K.; Suh, K. H.; Kim, D. Y. Synlett 2011, 1125–1128. doi:10.1055/s-0030-1259932
Return to citation in text: [1] -
Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2011, 52, 2356–2358. doi:10.1016/j.tetlet.2011.02.087
Return to citation in text: [1] -
Kang, S. H.; Kim, D. Y. Adv. Synth. Catal. 2010, 352, 2783–2786. doi:10.1002/adsc.201000515
Return to citation in text: [1] -
Moon, H. W.; Kim, D. Y. Tetrahedron Lett. 2010, 51, 2906–2908. doi:10.1016/j.tetlet.2010.03.105
Return to citation in text: [1] -
Kang, Y. K.; Kim, S. M.; Kim, D. Y. J. Am. Chem. Soc. 2010, 132, 11847–11849. doi:10.1021/ja103786c
Return to citation in text: [1] -
Kwon, B. K.; Kim, S. M.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 759–761. doi:10.1016/j.jfluchem.2009.06.002
Return to citation in text: [1] -
Oh, Y.; Kim, S. M.; Kim, D. Y. Tetrahedron Lett. 2009, 50, 4674–4676. doi:10.1016/j.tetlet.2009.06.003
Return to citation in text: [1] -
Lee, H. J.; Kang, S. H.; Kim, D. Y. Synlett 2011, 1559–1562. doi:10.1055/s-0030-1260770
Return to citation in text: [1] -
Yoon, S. J.; Kang, Y. K.; Kim, D. Y. Synlett 2011, 420–424. doi:10.1055/s-0030-1259319
Return to citation in text: [1] -
Kim, S. M.; Lee, J. H.; Kim, D. Y. Synlett 2008, 2659–2662. doi:10.1055/s-0028-1083510
Return to citation in text: [1]
| 1. | de Andrade-Neto, V. F.; Goulart, M. O. F.; da Silva Filho, J. F.; da Silva, M. J.; Pinto, M. C. F. R.; Pinto, A. V.; Zalis, M. G.; Carvalho, L. H.; Krettli, A. U. Bioorg. Med. Chem. Lett. 2004, 14, 1145–1149. doi:10.1016/j.bmcl.2003.12.069 |
| 2. | Tandon, V. K.; Yadav, D. B.; Singh, R. V.; Chaturvedi, A. K.; Shukla, P. K. Bioorg. Med. Chem. Lett. 2005, 15, 5324–5328. doi:10.1016/j.bmcl.2005.08.032 |
| 3. | Glänzel, M.; Bültmann, R.; Starke, K.; Frahm, A. W. Eur. J. Med. Chem. 2005, 40, 1262–1276. doi:10.1016/j.ejmech.2005.07.007 |
| 4. | Gomez-Monterrey, I.; Santelli, G.; Campiglia, P.; Califano, D.; Falasconi, F.; Pisano, C.; Vesci, L.; Lama, T.; Grieco, P.; Novellino, E. J. Med. Chem. 2005, 48, 1152–1157. doi:10.1021/jm0408565 |
| 13. | Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171–196. doi:10.1055/s-2001-10803 |
| 14. | Berner, O. M.; Tedeschi, L.; Enders, D. Eur. J. Org. Chem. 2002, 1877–1894. doi:10.1002/1099-0690(200206)2002:12<1877::AID-EJOC1877>3.0.CO;2-U |
| 15. | Christoffers, J.; Baro, A. Angew. Chem., Int. Ed. 2003, 42, 1688–1690. doi:10.1002/anie.200201614 |
| 10. | Corey, E. J.; Guzman-Perez, A. Angew. Chem., Int. Ed. 1998, 37, 388–401. doi:10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V |
| 11. | Christoffers, J.; Mann, A. Angew. Chem., Int. Ed. 2001, 40, 4591–4597. doi:10.1002/1521-3773(20011217)40:24<4591::AID-ANIE4591>3.0.CO;2-V |
| 34. | Zhou, W.-M.; Liu, H.; Du, D.-M. Org. Lett. 2008, 10, 2817–2820. doi:10.1021/ol800945e |
| 35. | Wu, R.; Chang, X.; Lu, A.; Wang, Y.; Wu, G.; Song, H.; Zhou, Z.; Tang, C. Chem. Commun. 2011, 47, 5034–5036. doi:10.1039/c1cc10797f |
| 36. | Yang, W.; Du, D.-M. Adv. Synth. Catal. 2011, 353, 1241–1246. doi:10.1002/adsc.201000981 |
| 5. | Gomez-Monterrey, I.; Campiglia, P.; Carotenuto, A.; Califano, D.; Pisano, C.; Vesci, L.; Lama, T.; Bertamino, A.; Sala, M.; Mazzella di Bosco, A.; Grieco, P.; Novellino, E. J. Med. Chem. 2007, 50, 1787–1798. doi:10.1021/jm0612158 |
| 6. | Castellano, S.; Bertamino, A.; Gomez-Monterrey, I.; Santoriello, M.; Grieco, P.; Campiglia, P.; Sbardella, G.; Novellino, E. Tetrahedron Lett. 2008, 49, 583–585. doi:10.1016/j.tetlet.2007.11.148 |
| 7. | Hsin, L.-W.; Wang, H.-P.; Kao, P.-H.; Lee, O.; Chen, W.-R.; Chen, H.-W.; Guh, J.-H.; Chan, Y.-L.; His, C.-P.; Yang, M.-S.; Li, T.-K.; Lee, C.-H. Bioorg. Med. Chem. 2008, 16, 1006–1014. doi:10.1016/j.bmc.2007.10.012 |
| 8. | Wei, P.; Zhang, X.; Tu, S.; Yan, S.; Ying, H.; Ouyang, P. Bioorg. Med. Chem. Lett. 2009, 19, 828–830. doi:10.1016/j.bmcl.2008.12.006 |
| 9. | Zhang, G.; Wang, Y.; Zhang, W.; Xu, X.; Zhong, A.; Xu, D. Eur. J. Org. Chem. 2011, 2142–2147. doi:10.1002/ejoc.201001570 |
| 34. | Zhou, W.-M.; Liu, H.; Du, D.-M. Org. Lett. 2008, 10, 2817–2820. doi:10.1021/ol800945e |
| 35. | Wu, R.; Chang, X.; Lu, A.; Wang, Y.; Wu, G.; Song, H.; Zhou, Z.; Tang, C. Chem. Commun. 2011, 47, 5034–5036. doi:10.1039/c1cc10797f |
| 36. | Yang, W.; Du, D.-M. Adv. Synth. Catal. 2011, 353, 1241–1246. doi:10.1002/adsc.201000981 |
| 43. | Kwon, B. K.; Kim, S. M.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 759–761. doi:10.1016/j.jfluchem.2009.06.002 |
| 44. | Oh, Y.; Kim, S. M.; Kim, D. Y. Tetrahedron Lett. 2009, 50, 4674–4676. doi:10.1016/j.tetlet.2009.06.003 |
| 27. | Hamashima, Y.; Hotta, D.; Sodeoka, M. J. Am. Chem. Soc. 2002, 124, 11240–11241. doi:10.1021/ja027075i |
| 28. | Wu, F.; Li, H.; Hong, R.; Deng, L. Angew. Chem., Int. Ed. 2006, 45, 947–950. doi:10.1002/anie.200502658 |
| 29. | Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2006, 47, 4565–4568. doi:10.1016/j.tetlet.2006.05.003 |
| 30. | Bartoli, G.; Bosco, M.; Carlone, A.; Cavalli, A.; Locatelli, M.; Mazzanti, A.; Ricci, P.; Sambri, L.; Melchiorre, P. Angew. Chem., Int. Ed. 2006, 45, 4966–4970. doi:10.1002/anie.200600370 |
| 31. | Rigby, C. L.; Dixon, D. J. Chem. Commun. 2008, 3798–3800. doi:10.1039/B805233F |
| 32. | Jung, S. H.; Kim, D. Y. Tetrahedron Lett. 2008, 49, 5527–5530. doi:10.1016/j.tetlet.2008.07.041 |
| 33. | Capuzzi, M.; Perdicchia, D.; Jørgensen, K. A. Chem.–Eur. J. 2008, 14, 128–135. doi:10.1002/chem.200701317 |
| 45. | Lee, H. J.; Kang, S. H.; Kim, D. Y. Synlett 2011, 1559–1562. doi:10.1055/s-0030-1260770 |
| 46. | Yoon, S. J.; Kang, Y. K.; Kim, D. Y. Synlett 2011, 420–424. doi:10.1055/s-0030-1259319 |
| 47. | Kim, S. M.; Lee, J. H.; Kim, D. Y. Synlett 2008, 2659–2662. doi:10.1055/s-0028-1083510 |
| 23. | Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: New York, 2001. |
| 24. | Calderari, G.; Seebach, D. Helv. Chim. Acta 1985, 68, 1592–1604. doi:10.1002/hlca.19850680611 |
| 25. | Ballini, R.; Petrini, M. Tetrahedron 2004, 60, 1017–1047. doi:10.1016/j.tet.2003.11.016 |
| 26. | Czekelius, C.; Carreira, E. M. Angew. Chem., Int. Ed. 2005, 44, 612–615. doi:10.1002/anie.200461879 |
| 16. | Connon, S. J. Angew. Chem., Int. Ed. 2006, 45, 3909–3912. doi:10.1002/anie.200600529 |
| 17. | Tylor, M. S.; Jacobson, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132 |
| 18. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 19. | Yu, X.; Wang, W. Chem.–Asian J. 2008, 3, 516–532. doi:10.1002/asia.200700415 |
| 20. | Connon, S. J. Synlett 2009, 354–376. doi:10.1055/s-0028-1087557 |
| 21. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 22. | Almaşi, D.; Alonso, D. A.; Nájera, D. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 37. | Kang, Y. K.; Kwon, B. K.; Mang, J. Y.; Kim, D. Y. Tetrahedron Lett. 2011, 52, 3247–3249. doi:10.1016/j.tetlet.2011.04.084 |
| 38. | Kang, Y. K.; Suh, K. H.; Kim, D. Y. Synlett 2011, 1125–1128. doi:10.1055/s-0030-1259932 |
| 39. | Kang, Y. K.; Kim, D. Y. Tetrahedron Lett. 2011, 52, 2356–2358. doi:10.1016/j.tetlet.2011.02.087 |
| 40. | Kang, S. H.; Kim, D. Y. Adv. Synth. Catal. 2010, 352, 2783–2786. doi:10.1002/adsc.201000515 |
| 41. | Moon, H. W.; Kim, D. Y. Tetrahedron Lett. 2010, 51, 2906–2908. doi:10.1016/j.tetlet.2010.03.105 |
| 42. | Kang, Y. K.; Kim, S. M.; Kim, D. Y. J. Am. Chem. Soc. 2010, 132, 11847–11849. doi:10.1021/ja103786c |
© 2012 Woo and Kim; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)