Abstract
In the context of synthetic efforts targeting the alkaloid lyconadin A, scalemic epoxide 25 was prepared by a highly stereoselective sequence involving a Myers alkylation and a Shi epoxidation. Ring-opening of this epoxide with a vinylcopper complex afforded alcohol 26 instead of the expected product 27. An unusual Lewis acid promoted Payne rearrangement of an α-trityloxy epoxide is proposed to account for this outcome.

Graphical Abstract
Introduction
Lyconadin A (1, Figure 1) was isolated from the club moss Lycopodium complanatum in 2001 by Kobayashi and co-workers [1]. Subsequent to this discovery, lyconadins B–F were isolated and characterized [2-4]. Biological assays revealed that 1 exhibits cytotoxicity against murine lymphoma L1210 and human epidermoid carcinoma KB cells (IC50 = 0.46 μg/mL and 1.7 μg/mL, respectively) [1]. Moreover, 1 has been shown to promote nerve growth factor biosynthesis in 1321N1 human astrocytoma cells [2]. In addition to its interesting bioactivity, lyconadin A presents a significant synthetic challenge by virtue of its unique pentacyclic skeleton, which contains six stereocenters and a pyridone ring. It is therefore not surprising that 1 has attracted the attention of the organic synthesis community. The first total synthesis of lyconadin A was reported in 2007 by Smith and Beshore [5,6], and efforts from the Sarpong [7,8] and Fukuyama [9,10] groups have also culminated in the construction of 1.
![[1860-5397-9-132-1]](/bjoc/content/figures/1860-5397-9-132-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our initial interest in lyconadin A was sparked by recognition that a 7-exo–6-exo cyclization cascade would efficiently furnish its bicyclo[5.4.0]undecane system, which is shown in bold in Figure 1. Subsequent to this observation, we performed model studies that demonstrated the viability of highly stereoselective 7-exo-trig acyl radical–6-exo-trig alkyl radical cyclizations as a means of preparing bicyclo[5.4.0]undecanes fused to aromatic rings [11]. Then, we devised an annulation protocol inspired by the work of Donohoe and co-workers [12,13] that provided access to substituted pyridones of the type found in 1 from thioester precursors [14]. Based on these encouraging results, we decided to target lyconadin A for synthesis. Herein, we provide an account of our studies directed toward the construction of this alkaloid. Specifically, we describe our efforts to prepare advanced intermediates that could be employed in the aforementioned pyridone annulation and tandem radical cyclization processes. In the course of this work, we discovered an unusual Payne-like rearrangement process that occurred in preference to the ring-opening of a hindered epoxide.
Results and Discussion
Our retrosynthetic analysis of lyconadin A is shown in Scheme 1. We reasoned that 1 could be formed by an alkylation cascade triggered by exposure of trimesylate 2 or a related electrophile to ammonia. A sequential alkylation process would serve as a viable alternative in the event of problems with this approach. In turn, cis-fused trimesylate 2 could be derived from trans-fused tricyclic ketone 3 by epimerization and standard functional-group manipulations. Based on the aforementioned model study [11], 7-exo–6-exo tandem radical cyclization of phenyl selenoester 4 was expected to produce ketone 3. Disassembly of the pyridone moiety of 4 according to our annulation protocol [14] revealed thioester 5 as a suitable precursor. We believed that this compound could be prepared from alkene 6 in two consecutive epoxidation–ring-opening sequences involving vinyl nucleophiles. We anticipated that a chiral catalyst such as one of the ketones developed by Shi and co-workers [15-18] would control the stereochemistry of the epoxidation of 6. Presumably, the identity of the protecting groups on this substrate (i.e., R2 and R3) would be critical to the success of the reaction. After formation of the epoxide, the bulky trityl ether was envisioned to direct the subsequent ring-opening to the distal carbon [19-22]. Alkene 6 would ultimately be formed by a Myers alkylation [23] of (+)-pseudoephedrine derived amide 7 with allylic iodide 8.
![[1860-5397-9-132-i1]](/bjoc/content/inline/1860-5397-9-132-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The initial epoxidation substrate of type 6 that we targeted possessed benzyl and TBDPS ethers as the protecting groups. First, allylic iodide 8 was synthesized by iodination of the mesylate derived from known alcohol 9 [24] (Scheme 2). Then, coupling of methyl γ-hydroxybutyrate (10) [25] with lithiated (+)-pseudoephedrine afforded amide 11 in excellent yield. Selective silylation of the primary alcohol of 11 delivered substrate 12. Alkylation of the enolate derived from 12 with 8 according to the Myers protocol [23] furnished adduct 13 in very high yield. Although not measured directly, the dr of this compound was assumed to be very high (i.e., ≥95:5) based on the results of an alkylation conducted on a very similar substrate (see below). The configuration of the newly formed stereocenter of 13 was assigned based on the established stereochemical course of the Myers alkylation [23]. Finally, reductive removal of the chiral auxiliary with lithium amidotrihydroborate [26] produced alcohol 14, and benzylation yielded triether 15.
![[1860-5397-9-132-i2]](/bjoc/content/inline/1860-5397-9-132-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Asymmetric epoxidation of alkene 15 was somewhat sluggish and required superstoichiometric amounts of Shi’s fructose-derived ketone 16 [27]. The resulting epoxide 17 was produced in moderate yield but excellent (<95:5) diastereomeric ratio (Scheme 3). The epoxide stereochemistry was assigned based on the reported outcomes of epoxidations mediated by 16 [27]. Epoxide 17 was then subjected to ring-opening reactions with vinyl Grignard reagents in the presence of various copper salts. Surprisingly, only trace amounts of the desired product were detected, with recovered starting material and multiple byproducts typically comprising the majority of the mass balance. Although not investigated in detail, analysis of these reactions by 1H NMR and mass spectrometry indicated that partial debenzylation was occurring. Accordingly, we decided to replace the benzyl ether with a 2-naphthylmethyl (NAP) ether [28]. Unfortunately, attempted protection of alcohol 14 produced varying yields of triether 19 along with byproducts derived from migration and/or scission of the TBDPS ether (Scheme 4). Modification of the reaction conditions failed to suppress the deleterious silyl migration and cleavage.
![[1860-5397-9-132-i3]](/bjoc/content/inline/1860-5397-9-132-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis and attempted ring-opening of epoxide 17.
Scheme 3: Synthesis and attempted ring-opening of epoxide 17.
![[1860-5397-9-132-i4]](/bjoc/content/inline/1860-5397-9-132-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Attempted protection of 14 and silyl migration.
Scheme 4: Attempted protection of 14 and silyl migration.
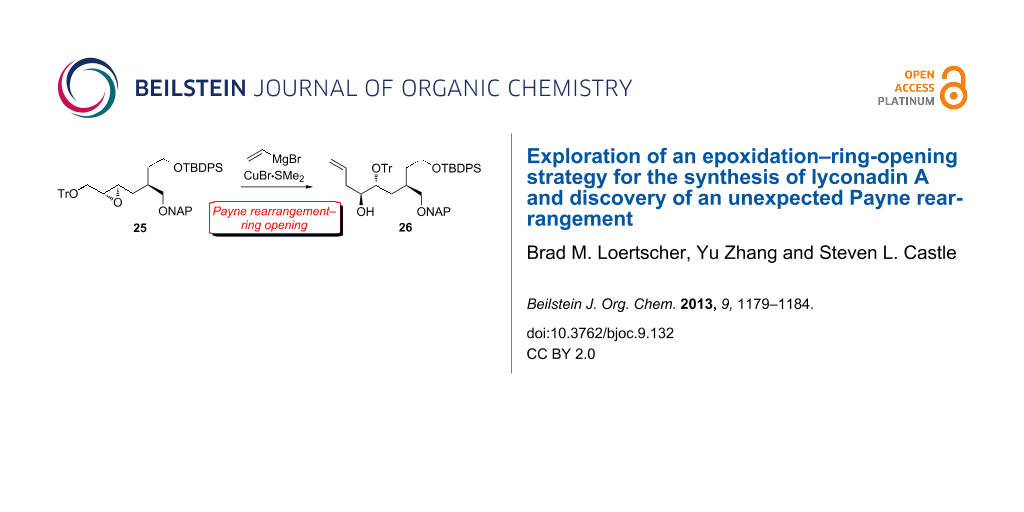
With the hope that a more robust silyl ether would not migrate, we installed a TIPS group on alcohol 11 (Scheme 5). Gratifyingly, TIPS-protected amide 20 was alkylated by 8 in the same yield as TBDPS-protected amide 12. Reductive removal of the chiral auxiliary furnished alcohol 22 in 91% yield. Fortunately, naphthylmethylation of 22 was achieved without migration of the TIPS group. Surprisingly, Shi epoxidation of the NAP ether derivative of 22 was low-yielding (<10%), and ring-opening of the resulting epoxide did not proceed. These results prompted us to swap the bulky TIPS moiety for a smaller TBDPS group, and triether 24 was obtained uneventfully in 77% overall yield from 22. Notably, the high (94%) ee of 24 as established by chiral HPLC analysis demonstrated that the Myers alkylation of 20 had proceeded with excellent diastereoselectivity. Then, we were pleased to find that Shi epoxidation of 24 provided 25 in reasonable (72%) yield and high (>95:5) dr. After considerable experimentation, we discovered that CuBr•Me2S [29] in conjunction with vinylmagnesium bromide was uniquely effective at facilitating the ring-opening of 25. However, careful inspection of the 1H NMR spectrum revealed the presence of one less hydrogen atom than expected in the 3–4 ppm region and one more hydrogen atom than expected in the 1–2 ppm region. Clearly, neither the anticipated product 27 nor the regioisomer derived from attack at the more hindered epoxide carbon had been generated. Instead, the NMR data were consistent with the formation of a different regioisomer, tentatively identified as alcohol 26, which had been produced in good yield.
![[1860-5397-9-132-i5]](/bjoc/content/inline/1860-5397-9-132-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis and ring-opening rearrangement of epoxide 25.
Scheme 5: Synthesis and ring-opening rearrangement of epoxide 25.
Presumably, the extremely hindered nature of internal epoxide 25 precluded its direct ring-opening, allowing alcohol 26 to form by means of a Payne rearrangement [30]. A possible mechanistic pathway for this transformation is given in Scheme 6. Coordination of a Lewis acid (likely a copper or magnesium species) to the trityl ether moiety of 25 could promote migration of the trityl group [31,32] to the epoxide, generating intermediate A. Payne rearrangement of A would then furnish epoxide B. Finally, attack of the vinylcopper complex [29] at the less-hindered carbon of the epoxide would provide 26. Acid- and Lewis acid promoted Payne rearrangements of epoxy alcohols [33,34] and epoxy methyl ethers [35] have been described, but we are unaware of any prior reports of Payne rearrangements of the bulkier epoxy trityl ethers. However, previous observations of trityl migration [31,32], although rare, do lend support to our mechanistic proposal. The NMR data for 26, while strongly supportive of the carbon backbone as drawn, do not permit an unambiguous assignment of the trityl ether to the C4 or C5 oxygen atom. An alternative pathway to this carbon skeleton involving a Payne rearrangement without trityl migration can also be envisioned, and under this scenario, the trityl ether would be located at C4 rather than C5. This possibility cannot be ruled out, but it would require opening of an activated epoxonium species at the less-substituted carbon instead of the more-substituted carbon as is typically observed. Thus, we favor the mechanism shown in Scheme 6.
![[1860-5397-9-132-i6]](/bjoc/content/inline/1860-5397-9-132-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism for generation of alcohol 26.
Scheme 6: Proposed mechanism for generation of alcohol 26.
To provide additional evidence for the structure of 26, this compound was converted into epoxide 29 as outlined in Scheme 7. Selective detritylation was accomplished by exposure to BCl3 at low temperature [36]. Camphorsulfonic acid was also effective for this transformation, although lengthy reaction times were required. Treatment of the resulting diol 28 with 2,4,6-triisopropylbenzenesulfonyl imidazole (TrisIm) effected regioselective sulfonylation (presumably of the less-hindered homoallylic alcohol, although this cannot be known for sure) followed by cyclization [37], delivering a single trans-disubstituted epoxide 29 of uncertain absolute stereochemistry in good yield. Examination of the 1H NMR spectrum of 29 clearly demonstrated that a disubstituted epoxide had been generated. Alcohol 27, or the aforementioned regioisomer that would have resulted from ring-opening of epoxide 25 at the more hindered carbon, would have afforded terminal epoxide 30 or oxetane 31, respectively, when subjected to this two-step sequence. While these observations do not shed light on the location of the trityl ether in 26, they do provide compelling evidence that the carbon backbone of this compound is correct as drawn and is produced by a Payne rearrangement of some type.
![[1860-5397-9-132-i7]](/bjoc/content/inline/1860-5397-9-132-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of epoxide 29 from alcohol 26 (asterisks indicate relative but not absolute stereochemistry).
Scheme 7: Synthesis of epoxide 29 from alcohol 26 (asterisks indicate relative but not absolute stereochemist...
Conclusion
In the context of synthetic efforts targeting the polycyclic alkaloid lyconadin A, we prepared scalemic epoxide 25. A Myers alkylation and a reagent-controlled Shi epoxidation were used to construct this compound in a highly stereoselective fashion. The bulky trityl group of 25 was intended to serve as a means of directing a ring-opening reaction to the distal carbon of the epoxide [19-22]. However, an unanticipated Lewis acid promoted Payne rearrangement intervened, producing alcohol 26 instead of the expected regioisomer 27. We believe that the extremely hindered nature of epoxide 25 prevented the desired ring-opening process, thereby enabling the unusual rearrangement to proceed. Conceivably, future studies of the scope and limitations of Lewis acid promoted Payne rearrangement–ring-opening cascades could establish their utility in organic synthesis.
Supporting Information
| Supporting Information File 1: Name: Experimental procedures and characterization data for all new compounds. | ||
| Format: PDF | Size: 322.9 KB | Download |
Acknowledgements
We thank the National Science Foundation (CHE-716991) and Brigham Young University (Roland K. Robins Fellowship to B.M.L.) for financial support. We also thank Nathan C. Wilde for assistance with the preparation of starting materials and Professor Douglass F. Taber for helpful discussions.
References
-
Kobayashi, J.; Hirasawa, Y.; Yoshida, N.; Morita, H. J. Org. Chem. 2001, 66, 5901–5904. doi:10.1021/jo0103874
Return to citation in text: [1] [2] -
Ishiuchi, K.; Kubota, T.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2006, 14, 5995–6000. doi:10.1016/j.bmc.2006.05.028
Return to citation in text: [1] [2] -
Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Kobayashi, J. Tetrahedron Lett. 2011, 52, 289–292. doi:10.1016/j.tetlet.2010.11.024
Return to citation in text: [1] -
Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Mori, K.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2011, 19, 749–753. doi:10.1016/j.bmc.2010.12.025
Return to citation in text: [1] -
Beshore, D. C.; Smith, A. B., III. J. Am. Chem. Soc. 2007, 129, 4148–4149. doi:10.1021/ja070336+
Return to citation in text: [1] -
Beshore, D. C.; Smith, A. B., III. J. Am. Chem. Soc. 2008, 130, 13778–13789. doi:10.1021/ja804939r
Return to citation in text: [1] -
Bisai, A.; West, S. P.; Sarpong, R. J. Am. Chem. Soc. 2008, 130, 7222–7223. doi:10.1021/ja8028069
Return to citation in text: [1] -
West, S. P.; Bisai, A.; Lim, A. D.; Narayan, R. R.; Sarpong, R. J. Am. Chem. Soc. 2009, 191, 11187–11194. doi:10.1021/ja903868n
Return to citation in text: [1] -
Nishimura, T.; Unni, A. K.; Yokoshima, S.; Fukuyama, T. J. Am. Chem. Soc. 2011, 133, 418–419. doi:10.1021/ja109516f
Return to citation in text: [1] -
Nishimura, T.; Unni, A. K.; Yokoshima, S.; Fukuyama, T. J. Am. Chem. Soc. 2013, 135, 3243–3247. doi:10.1021/ja312065m
Return to citation in text: [1] -
Grant, S. W.; Zhu, K.; Zhang, Y.; Castle, S. L. Org. Lett. 2006, 8, 1867–1870. doi:10.1021/ol0604264
Return to citation in text: [1] [2] -
Donohoe, T. J.; Fishlock, L. P.; Procopiou, P. A. Org. Lett. 2008, 10, 285–288. doi:10.1021/ol702684d
Return to citation in text: [1] -
Donohoe, T. J.; Fishlock, L. P.; Procopiou, P. A. Synthesis 2008, 2665–2667. doi:10.1055/s-2008-1067180
Return to citation in text: [1] -
Zhang, Y.; Loertscher, B. M.; Castle, S. L. Tetrahedron 2009, 65, 6584–6590. doi:10.1016/j.tet.2009.06.006
Return to citation in text: [1] [2] -
Shi, Y. Acc. Chem. Res. 2004, 37, 488–496. doi:10.1021/ar030063x
Return to citation in text: [1] -
Frohn, M.; Shi, Y. Synthesis 2000, 1979–2000. doi:10.1055/s-2000-8715
Return to citation in text: [1] -
Wong, O. A.; Shi, Y. Top. Curr. Chem. 2010, 291, 201–232. doi:10.1007/128_2008_16
Return to citation in text: [1] -
Wong, O. A.; Shi, Y. Chem. Rev. 2008, 108, 3958–3987. doi:10.1021/cr068367v
Return to citation in text: [1] -
Taber, D. F.; Green, J. H.; Geremia, J. M. J. Org. Chem. 1997, 62, 9342–9344. doi:10.1021/jo9710386
Return to citation in text: [1] [2] -
Yang, C.-G.; Wang, J.; Jiang, B. Tetrahedron Lett. 2002, 43, 1063–1066. doi:10.1016/S0040-4039(01)02331-0
Return to citation in text: [1] [2] -
Watanabe, B.; Yamamoto, S.; Sasaki, K.; Nakagawa, Y.; Miyagawa, H. Tetrahedron Lett. 2004, 45, 2767–2769. doi:10.1016/j.tetlet.2004.02.033
Return to citation in text: [1] [2] -
Roulland, E.; Ermolenko, M. S. Org. Lett. 2005, 7, 2225–2228. doi:10.1021/ol050588k
Return to citation in text: [1] [2] -
Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f
Return to citation in text: [1] [2] [3] -
Hernández, A.-I.; Balzarini, J.; Karlsson, A.; Camarasa, M.-J.; Pérez-Pérez, M.-J. J. Med. Chem. 2002, 45, 4254–4263. doi:10.1021/jm011128+
Return to citation in text: [1] -
Gannett, P. M.; Nagel, D. L.; Reilly, P. J.; Lawson, T.; Sharpe, J.; Toth, B. J. Org. Chem. 1988, 53, 1064–1071. doi:10.1021/jo00240a024
Return to citation in text: [1] -
Myers, A. G.; Yang, B. H.; Kopecky, D. J. Tetrahedron Lett. 1996, 37, 3623–3626. doi:10.1016/0040-4039(96)00652-1
Return to citation in text: [1] -
Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224–11235. doi:10.1021/ja972272g
Return to citation in text: [1] [2] -
Inoue, M.; Uehara, H.; Maruyama, M.; Hirama, M. Org. Lett. 2002, 4, 4551–4554. doi:10.1021/ol027105m
Return to citation in text: [1] -
Marfat, A.; McGuirk, P. R.; Helquist, P. J. Org. Chem. 1979, 44, 3888–3901. doi:10.1021/jo01336a029
Return to citation in text: [1] [2] -
Hanson, R. M. Org. React. 2002, 60, 1–156.
Return to citation in text: [1] -
Bartlett, P. A.; Green, F. R., III. J. Am. Chem. Soc. 1978, 100, 4858–4865. doi:10.1021/ja00483a036
Return to citation in text: [1] [2] -
Cabral, N. L. D.; Hoeltgebaum Thiessen, L. J.; Doboszewski, B. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 931–948. doi:10.1080/15257770802257846
Return to citation in text: [1] [2] -
Morrison, G. A.; Wilkinson, J. B. J. Chem. Soc., Perkin Trans. 1 1989, 2003–2007. doi:10.1039/P19890002003
Return to citation in text: [1] -
Wu, M. H.; Hansen, K. B.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2012–2014. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<2012::AID-ANIE2012>3.0.CO;2-H
Return to citation in text: [1] -
Morrison, G. A.; Wilkinson, J. B. J. Chem. Soc., Perkin Trans. 1 1990, 345–351. doi:10.1039/P19900000345
Return to citation in text: [1] -
Jones, G. B.; Hynd, G.; Wright, J. M.; Sharma, A. J. Org. Chem. 2000, 65, 263–265. doi:10.1021/jo9913255
Return to citation in text: [1] -
Smith, A. B., III; Doughty, V. A.; Sfouggatakis, C.; Bennett, C. S.; Koyanagi, J.; Takeuchi, M. Org. Lett. 2002, 4, 783–786. doi:10.1021/ol017273z
Return to citation in text: [1]
| 33. | Morrison, G. A.; Wilkinson, J. B. J. Chem. Soc., Perkin Trans. 1 1989, 2003–2007. doi:10.1039/P19890002003 |
| 34. | Wu, M. H.; Hansen, K. B.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2012–2014. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<2012::AID-ANIE2012>3.0.CO;2-H |
| 35. | Morrison, G. A.; Wilkinson, J. B. J. Chem. Soc., Perkin Trans. 1 1990, 345–351. doi:10.1039/P19900000345 |
| 31. | Bartlett, P. A.; Green, F. R., III. J. Am. Chem. Soc. 1978, 100, 4858–4865. doi:10.1021/ja00483a036 |
| 32. | Cabral, N. L. D.; Hoeltgebaum Thiessen, L. J.; Doboszewski, B. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 931–948. doi:10.1080/15257770802257846 |
| 1. | Kobayashi, J.; Hirasawa, Y.; Yoshida, N.; Morita, H. J. Org. Chem. 2001, 66, 5901–5904. doi:10.1021/jo0103874 |
| 5. | Beshore, D. C.; Smith, A. B., III. J. Am. Chem. Soc. 2007, 129, 4148–4149. doi:10.1021/ja070336+ |
| 6. | Beshore, D. C.; Smith, A. B., III. J. Am. Chem. Soc. 2008, 130, 13778–13789. doi:10.1021/ja804939r |
| 23. | Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f |
| 2. | Ishiuchi, K.; Kubota, T.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2006, 14, 5995–6000. doi:10.1016/j.bmc.2006.05.028 |
| 24. | Hernández, A.-I.; Balzarini, J.; Karlsson, A.; Camarasa, M.-J.; Pérez-Pérez, M.-J. J. Med. Chem. 2002, 45, 4254–4263. doi:10.1021/jm011128+ |
| 1. | Kobayashi, J.; Hirasawa, Y.; Yoshida, N.; Morita, H. J. Org. Chem. 2001, 66, 5901–5904. doi:10.1021/jo0103874 |
| 15. | Shi, Y. Acc. Chem. Res. 2004, 37, 488–496. doi:10.1021/ar030063x |
| 16. | Frohn, M.; Shi, Y. Synthesis 2000, 1979–2000. doi:10.1055/s-2000-8715 |
| 17. | Wong, O. A.; Shi, Y. Top. Curr. Chem. 2010, 291, 201–232. doi:10.1007/128_2008_16 |
| 18. | Wong, O. A.; Shi, Y. Chem. Rev. 2008, 108, 3958–3987. doi:10.1021/cr068367v |
| 2. | Ishiuchi, K.; Kubota, T.; Hoshino, T.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2006, 14, 5995–6000. doi:10.1016/j.bmc.2006.05.028 |
| 3. | Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Kobayashi, J. Tetrahedron Lett. 2011, 52, 289–292. doi:10.1016/j.tetlet.2010.11.024 |
| 4. | Ishiuchi, K.; Kubota, T.; Ishiyama, H.; Hayashi, S.; Shibata, T.; Mori, K.; Obara, Y.; Nakahata, N.; Kobayashi, J. Bioorg. Med. Chem. 2011, 19, 749–753. doi:10.1016/j.bmc.2010.12.025 |
| 19. | Taber, D. F.; Green, J. H.; Geremia, J. M. J. Org. Chem. 1997, 62, 9342–9344. doi:10.1021/jo9710386 |
| 20. | Yang, C.-G.; Wang, J.; Jiang, B. Tetrahedron Lett. 2002, 43, 1063–1066. doi:10.1016/S0040-4039(01)02331-0 |
| 21. | Watanabe, B.; Yamamoto, S.; Sasaki, K.; Nakagawa, Y.; Miyagawa, H. Tetrahedron Lett. 2004, 45, 2767–2769. doi:10.1016/j.tetlet.2004.02.033 |
| 22. | Roulland, E.; Ermolenko, M. S. Org. Lett. 2005, 7, 2225–2228. doi:10.1021/ol050588k |
| 12. | Donohoe, T. J.; Fishlock, L. P.; Procopiou, P. A. Org. Lett. 2008, 10, 285–288. doi:10.1021/ol702684d |
| 13. | Donohoe, T. J.; Fishlock, L. P.; Procopiou, P. A. Synthesis 2008, 2665–2667. doi:10.1055/s-2008-1067180 |
| 11. | Grant, S. W.; Zhu, K.; Zhang, Y.; Castle, S. L. Org. Lett. 2006, 8, 1867–1870. doi:10.1021/ol0604264 |
| 19. | Taber, D. F.; Green, J. H.; Geremia, J. M. J. Org. Chem. 1997, 62, 9342–9344. doi:10.1021/jo9710386 |
| 20. | Yang, C.-G.; Wang, J.; Jiang, B. Tetrahedron Lett. 2002, 43, 1063–1066. doi:10.1016/S0040-4039(01)02331-0 |
| 21. | Watanabe, B.; Yamamoto, S.; Sasaki, K.; Nakagawa, Y.; Miyagawa, H. Tetrahedron Lett. 2004, 45, 2767–2769. doi:10.1016/j.tetlet.2004.02.033 |
| 22. | Roulland, E.; Ermolenko, M. S. Org. Lett. 2005, 7, 2225–2228. doi:10.1021/ol050588k |
| 11. | Grant, S. W.; Zhu, K.; Zhang, Y.; Castle, S. L. Org. Lett. 2006, 8, 1867–1870. doi:10.1021/ol0604264 |
| 14. | Zhang, Y.; Loertscher, B. M.; Castle, S. L. Tetrahedron 2009, 65, 6584–6590. doi:10.1016/j.tet.2009.06.006 |
| 9. | Nishimura, T.; Unni, A. K.; Yokoshima, S.; Fukuyama, T. J. Am. Chem. Soc. 2011, 133, 418–419. doi:10.1021/ja109516f |
| 10. | Nishimura, T.; Unni, A. K.; Yokoshima, S.; Fukuyama, T. J. Am. Chem. Soc. 2013, 135, 3243–3247. doi:10.1021/ja312065m |
| 36. | Jones, G. B.; Hynd, G.; Wright, J. M.; Sharma, A. J. Org. Chem. 2000, 65, 263–265. doi:10.1021/jo9913255 |
| 7. | Bisai, A.; West, S. P.; Sarpong, R. J. Am. Chem. Soc. 2008, 130, 7222–7223. doi:10.1021/ja8028069 |
| 8. | West, S. P.; Bisai, A.; Lim, A. D.; Narayan, R. R.; Sarpong, R. J. Am. Chem. Soc. 2009, 191, 11187–11194. doi:10.1021/ja903868n |
| 14. | Zhang, Y.; Loertscher, B. M.; Castle, S. L. Tetrahedron 2009, 65, 6584–6590. doi:10.1016/j.tet.2009.06.006 |
| 37. | Smith, A. B., III; Doughty, V. A.; Sfouggatakis, C.; Bennett, C. S.; Koyanagi, J.; Takeuchi, M. Org. Lett. 2002, 4, 783–786. doi:10.1021/ol017273z |
| 23. | Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f |
| 25. | Gannett, P. M.; Nagel, D. L.; Reilly, P. J.; Lawson, T.; Sharpe, J.; Toth, B. J. Org. Chem. 1988, 53, 1064–1071. doi:10.1021/jo00240a024 |
| 23. | Myers, A. G.; Yang, B. H.; Chen, H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. J. Am. Chem. Soc. 1997, 119, 6496–6511. doi:10.1021/ja970402f |
| 31. | Bartlett, P. A.; Green, F. R., III. J. Am. Chem. Soc. 1978, 100, 4858–4865. doi:10.1021/ja00483a036 |
| 32. | Cabral, N. L. D.; Hoeltgebaum Thiessen, L. J.; Doboszewski, B. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 931–948. doi:10.1080/15257770802257846 |
| 29. | Marfat, A.; McGuirk, P. R.; Helquist, P. J. Org. Chem. 1979, 44, 3888–3901. doi:10.1021/jo01336a029 |
| 29. | Marfat, A.; McGuirk, P. R.; Helquist, P. J. Org. Chem. 1979, 44, 3888–3901. doi:10.1021/jo01336a029 |
| 27. | Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224–11235. doi:10.1021/ja972272g |
| 28. | Inoue, M.; Uehara, H.; Maruyama, M.; Hirama, M. Org. Lett. 2002, 4, 4551–4554. doi:10.1021/ol027105m |
| 26. | Myers, A. G.; Yang, B. H.; Kopecky, D. J. Tetrahedron Lett. 1996, 37, 3623–3626. doi:10.1016/0040-4039(96)00652-1 |
| 27. | Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224–11235. doi:10.1021/ja972272g |
© 2013 Loertscher et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)