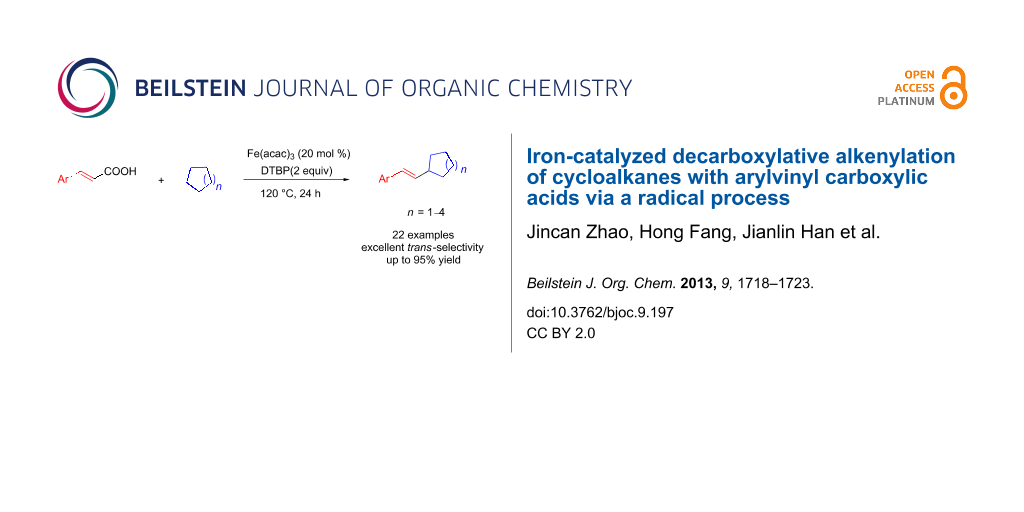
Abstract
A Fe(acac)3-catalyzed decarboxylative coupling of 2-(aryl)vinyl carboxylic acids with cycloalkanes was developed by using DTBP as an oxidant through a radical process. This reaction tolerates a wide range of substrates, and products are obtained in good to excellent yields (71–95%). The reaction also shows excellent stereoselectivity, and only trans-isomers are obtained.

Graphical Abstract
Introduction
Direct C–H functionalization has become one of the most useful and attractive tools in organic chemistry because it can construct carbon–carbon or carbon–heteroatom bonds in a highly atom economical manner [1-8]. Among all these C–H functionalization methods, the direct C(sp3)–H functionalization attracts particular attention due to its low reactivity and challenging activation [9-11]. In previous studies, numerous transition-metal-catalyzed processes, such as Pd [12-18], Cu [19-23], Ru [24-27], Rh [28-31], Co [32-34], Au [35,36], Ir [37-39], Fe [40-43] and other metals [44-47], have been developed for sp3 C–H activation reactions. Additionally, metal-free methodologies, which use TBHP, PhI(OAc)2, TBAI, I2 or Lewis/Brønsted acids, have also been employed for cross-dehydrogenative coupling reactions [48-57].
Owing to the general low reactivity of cycloalkane C(sp3)–H bonds, the direct alkenylation of cycloalkanes with high selectivity and stereospecificity remains a great challenge and attracted a lot of attention in the past years. In 1996, the Fuchs group described the alkenylation of cyclohexane by a radical reaction with vinyl triflone [58]. In 2003, the Yao group reported that styryl cycloalkanes were prepared based on a radical substitution of cyclohydrocarbon units to (E)-β-nitrostyrenes by using the radical initiator benzoyl peroxide [59]. Recently, the Liu group developed a copper-catalyzed decarboxylative coupling of vinylic carboxylic acids with simple alcohols and ethers in high yields. Cycloalkanes were also investigated in this catalytic system, though only moderate yields were obtained [60].
Several other groups have also found that Pd, Ag or Cu could catalyze the decarboxylative coupling of various aromatic, alkenyl, and alkynyl carboxylic acids [61-68]. Our group was surprised to find that low-cost Fe(acac)3 could catalyze the direct alkenylation of cyclohexane sp3 C–H bonds by decarboxylative couplings with high efficiency.
Results and Discussion
We initiated our investigation by reacting cinnamic acid (1a, 0.3 mmol) with cyclohexane (2a, 2 mL) in the presence of iron(II) chloride tetrahydrate (20 mol %) and 2.0 equiv of di-tert-butyl peroxide (DTBP) as the oxidant at 120 °C under nitrogen, which provided the expected (E)-(2-cyclohexylvinyl)benzene (3a), but in a moderate 54% yield (Table 1, entry 1). The use of aqueous TBHP as oxidant instead of DTBP reduced the yield to only 38% (Table 1, entry 2). With the help of 1,10-phenanthroline (30 mol %) as the ligand, the yield could be slightly improved to 68% (Table 1, entry 3). Iron(III) acetylacetonate provided a superior yield (91%) compared to the other Fe salts such as FeCl3, ferrocene, Fe2O3 and Fe3O4 tested (Table 1, entries 4–8). Application of other oxidants such as K2S2O8, H2O2 (30% aqueous solution) or TBPB did not afford any improvements (Table 1, entries 9–11). A decreased loading of Fe(acac)3 to 10 mol % or an increased amount of DTBP to 5.0 equiv and a lower temperature (110 °C), decreased the yield to 63%, 69% and 79%, respectively (Table 1, entries 12–14). The reaction did not proceed without the iron catalyst or DTBP (Table 1, entries 15 and 16).
Table 1: Optimization of typical reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-197-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | cat. (mol %) | oxidant | yield (%)b |
|---|---|---|---|
| 1 | FeCl2·4H2O (20) | DTBP | 54 |
| 2 | FeCl2·4H2O (20) | TBHP | 38 |
| 3 | FeCl2·4H2O (20) | DTBP | 68c |
| 4 | FeCl3 (20) | DTBP | Trace |
| 5 | Ferrocene (20) | DTBP | 74 |
| 6 | Fe2O3 (20) | DTBP | 78 |
| 7 | Fe3O4 (20) | DTBP | 80 |
| 8 | Fe(acac)3 (20) | DTBP | 91 |
| 9 | Fe(acac)3 (20) | K2S2O8 | N.D. |
| 10 | Fe(acac)3 (20) | H2O2d | 21 |
| 11 | Fe(acac)3 (20) | TBPB | 49 |
| 12 | Fe(acac)3 (10) | DTBPe | 63 |
| 13f | Fe(acac)3 (20) | DTBP | 69 |
| 14 | Fe(acac)3 (20)g | DTBP | 79 |
| 15 | Fe(acac)3 (20) | – | N.D. |
| 16 | – | DTBP | N.D. |
aCatalytic conditions: cinnamic acid (0.3 mmol), cyclohexane (2 mL), iron catalyst (20 mol %), oxidant (2.0 equiv), 120 °C, 24 h, N2. bIsolated yields based on cinnamic acid. cUsing 1,10-phenanthroline (30 mol %) as the ligand. d30% aqueous solution. e5 equiv. f12 h. g110 °C.
We then examined the substrate scope and limitation of the procedure by reacting cyclohexane with a variety of substituted cinnamic acid derivatives under the optimized conditions (Table 1, entry 8). As shown in Scheme 1, almost all of the tested substrates worked well in this reaction. Several substituents on the aromatic ring were tolerated and the position of these substituents showed almost no effect on the chemical yield. We also observed that electron-donating substituents, such as methyl or methoxy groups at any position of the ring, efficiently took part in the reaction with a slightly decreased yield in case of ortho-substituted products (Scheme 1, 3b–g). Furthermore, a reaction of 2,6-disubstituted cinnamic acid 1m lead to the expected product 3m, which was obtained in a lower yield due to steric hindrance. In addition, heteroaryl-substituted acrylic acids can also be efficiently converted under these conditions. This was shown by the reaction of 3-(thiophen-2-yl)acrylic acid (1p) with cyclohexane furnishing the product 3p in 89% yield. In general, the stereoselectivity of this reaction was excellent and only trans-isomers were obtained in all cases.
![[1860-5397-9-197-i1]](/bjoc/content/inline/1860-5397-9-197-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fe(acac)3-catalyzed alkenylation of cyclohexane. Catalytic conditions: cinnamic acid (1) (0.3 mmol), cyclohexane (2 mL), Fe(acac)3 (20 mol %), DTBP (2.0 equiv), 120 °C, 24 h, N2. Yields are isolated yields based on cinnamic acid.
Scheme 1: Fe(acac)3-catalyzed alkenylation of cyclohexane. Catalytic conditions: cinnamic acid (1) (0.3 mmol)...
Next, other cycloalkanes, including cyclopentane, cycloheptane and cyclooctane, were reacted with different cinnamic acids 1, giving products 4a–f in 83–95% chemical yield (Scheme 2). As already shown for cyclohexane as a substrate, the reaction of other cycloalkanes performs equally well with a variety of cinammic acid derivatives under these conditions. It is noteworthy, that the decarboxylative cross-coupling with cyclooctane showed a higher efficiency than with smaller cycloalkanes.
![[1860-5397-9-197-i2]](/bjoc/content/inline/1860-5397-9-197-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Fe(acac)3-catalyzed alkenylation of cyclopentan, cycloheptane and cyclooctane. Catalytic conditions: cinnamic acid (1) (0.3 mmol), cycloalkanes (2.0 mL), Fe(acac)3 (20 mol %), DTBP (2 equiv), 120 °C, 24 h, N2. Yields are isolated yields based on cinnamic acid.
Scheme 2: Fe(acac)3-catalyzed alkenylation of cyclopentan, cycloheptane and cyclooctane. Catalytic conditions...
Finally two control experiments were carried out to shed light on the reaction mechanism. Addition of the radical scavenger 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) or azobisisobutyronitrile (AIBN) completely inhibited the reaction, and almost no desired product was obtained. Based on these results and literature reports [69,70], a plausible mechanism for the radical oxidative coupling is illustrated in Scheme 3. At the beginning, Fe-catalyzed cleavage of DTBP by Fe(III) in the presence of cinnamic acid, gives tert-butoxy radical A, intermediate B and one acac. Next, a cyclohexane radical C is generated by the reaction between tert-butoxy radical A and cyclohexane. Subsequently, addition of cyclohexane radical C to the α-position of the double bond in B gives intermediate D. Finally, the radical intermediate D is oxidatively decarboxylated by Fe(III) to give product 3, Fe(II) and carbon dioxide. The Fe(III) catalyst is then reformed via DTBP oxidation [71].
![[1860-5397-9-197-i3]](/bjoc/content/inline/1860-5397-9-197-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: A plausible pathway for the reaction.
Scheme 3: A plausible pathway for the reaction.
Conclusion
In conclusion, an efficient procedure for the Fe(acac)3-catalyzed direct alkenylation of sp3 C–H bonds of cycloalkanes with DTBP as an oxidant has been reported. This method provides a useful strategy for the stereospecific synthesis of substituted E-alkenes. Various cinnamic acids and cycloalkanes are well-tolerated in this catalytic system with good to excellent chemical yields. The mechanism of this reaction has also been studied, and a radical mechanism was proposed. Further studies on the alkenylation of other sp3 C–H substrates are currently investigated in our laboratory.
Experimental
General procedure for the iron-catalyzed decarboxylative alkenylation of cycloalkanes: To a Schlenk tube equipped with a magnetic stir bar were added Fe(acac)3 (21.2 mg, 0.06 mmol) and cinnamic acid (0.3 mmol) under a nitrogen atmosphere. Cycloalkane (2.0 mL, 15–25 mmol) and DTBP (di-tert-butyl peroxide, 0.6 mmol, 113 μL) were added under a nitrogen atmosphere and the resulting reaction mixture was stirred at 120 °C for 24 h. After cooling to room temperature and removal of volatiles, the products were isolated by flash column chromatography (PE).
Supporting Information
| Supporting Information File 1: Experimental details and spectral data. | ||
| Format: PDF | Size: 904.7 KB | Download |
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21102071) and the Fundamental Research Funds for the Central Universities (No. 1107020522 and No. 1082020502). The Jiangsu 333 program (for Pan) and Changzhou Jin-Feng-Huang program (for Han) are also acknowledged.
References
-
Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. doi:10.1002/anie.200806273
Return to citation in text: [1] -
Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074–1086. doi:10.1021/ar9000058
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n
Return to citation in text: [1] -
Coperet, C. Chem. Rev. 2010, 110, 656–680. doi:10.1021/cr900122p
Return to citation in text: [1] -
Werner, H. Angew. Chem., Int. Ed. 2010, 49, 4714–4728. doi:10.1002/anie.201000306
Return to citation in text: [1] -
Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293–1314. doi:10.1021/cr100198w
Return to citation in text: [1] -
Campbell, A. N.; Stahl, S. S. Acc. Chem. Res. 2012, 45, 851–863. doi:10.1021/ar2002045
Return to citation in text: [1] -
Kozhushkov, S. I.; Ackermann, L. Chem. Sci. 2013, 4, 886–896. doi:10.1039/c2sc21524a
Return to citation in text: [1] -
Chatani, N.; Asaumi, T.; Yorimitsu, S.; Ikeda, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 10935–10941. doi:10.1021/ja011540e
Return to citation in text: [1] -
Jiang, X. F.; Shen, M.; Tang, Y.; Li, C. Tetrahedron Lett. 2005, 46, 487–489. doi:10.1016/j.tetlet.2004.11.113
Return to citation in text: [1] -
Song, C.-X.; Cai, G.-X.; Farrell, T. R.; Jiang, Z.-P.; Li, H.; Gan, L.-B.; Shi, Z.-J. Chem. Commun. 2009, 6002–6004. doi:10.1039/b911031c
Return to citation in text: [1] -
Guin, S.; Rout, S. K.; Banerjee, A.; Nandi, S.; Patel, B. K. Org. Lett. 2012, 14, 5294–5297. doi:10.1021/ol302438z
Return to citation in text: [1] -
Rousseaux, S.; Liégault, B.; Fagnou, K. Chem. Sci. 2012, 3, 244–248. doi:10.1039/c1sc00458a
Return to citation in text: [1] -
Solé, D.; Mariani, F.; Fernández, I.; Sierra, M. A. J. Org. Chem. 2012, 77, 10272–10284. doi:10.1021/jo301924e
Return to citation in text: [1] -
Wasa, M.; Chan, K. S. L.; Zhang, X.-G.; He, J.; Miura, M.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 18570–18572. doi:10.1021/ja309325e
Return to citation in text: [1] -
Yin, Z.; Sun, P. J. Org. Chem. 2012, 77, 11339–11344. doi:10.1021/jo302125h
Return to citation in text: [1] -
Saget, T.; Perez, D.; Cramer, N. Org. Lett. 2013, 15, 1354–1357. doi:10.1021/ol400380y
Return to citation in text: [1] -
Zhang, S.-Y.; He, G.; Nack, W. A.; Zhao, Y.; Li, Q.; Chen, G. J. Am. Chem. Soc. 2013, 135, 2124–2127. doi:10.1021/ja312277g
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763
Return to citation in text: [1] -
Xie, J.; Huang, Z.-Z. Angew. Chem., Int. Ed. 2010, 49, 10181–10185. doi:10.1002/anie.201004940
Return to citation in text: [1] -
Yang, F.; Li, J.; Xie, J.; Huang, Z.-Z. Org. Lett. 2010, 12, 5214–5217. doi:10.1021/ol102252n
Return to citation in text: [1] -
Rout, S. K.; Guin, S.; Ghara, K. K.; Banerjee, A.; Patel, B. K. Org. Lett. 2012, 14, 3982–3985. doi:10.1021/ol301756y
Return to citation in text: [1] -
Xia, R.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Org. Lett. 2012, 14, 5546–5549. doi:10.1021/ol302640e
Return to citation in text: [1] -
Pastine, S. J.; Gribkov, D. V.; Sames, D. J. Am. Chem. Soc. 2006, 128, 14220–14221. doi:10.1021/ja064481j
Return to citation in text: [1] -
Rankin, M. A.; Schatte, G.; McDonald, R.; Stradiotto, M. J. Am. Chem. Soc. 2007, 129, 6390–6391. doi:10.1021/ja071684e
Return to citation in text: [1] -
Deng, G.; Zhao, L.; Li, C.-J. Angew. Chem., Int. Ed. 2008, 47, 6278–6282. doi:10.1002/anie.200801544
Return to citation in text: [1] -
Wang, M.-Z.; Zhou, C.-Y.; Wong, M.-K.; Che, C.-M. Chem.–Eur. J. 2010, 16, 5723–5735. doi:10.1002/chem.200902387
Return to citation in text: [1] -
Shi, L.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M.; Fan, C.-A.; Zhao, Y.-M.; Xia, W.-J. J. Am. Chem. Soc. 2005, 127, 10836–10837. doi:10.1021/ja0528331
Return to citation in text: [1] -
Jo, E.-A.; Lee, J.-H.; Jun, C.-H. Chem. Commun. 2008, 5779–5781. doi:10.1039/b814166e
Return to citation in text: [1] -
Rakshit, S.; Patureau, F. W.; Glorius, F. J. Am. Chem. Soc. 2010, 132, 9585–9587. doi:10.1021/ja104305s
Return to citation in text: [1] -
Kuninobu, Y.; Nakahara, T.; Takeshima, H.; Takai, K. Org. Lett. 2013, 15, 426–428. doi:10.1021/ol303353m
Return to citation in text: [1] -
Harden, J. D.; Ruppel, J. V.; Gao, G.-Y.; Zhang, X. P. Chem. Commun. 2007, 4644–4646. doi:10.1039/b710677g
Return to citation in text: [1] -
Hung-Low, F.; Krogman, J. P.; Tye, J. W.; Bradley, C. A. Chem. Commun. 2012, 48, 368–370. doi:10.1039/c1cc15458c
Return to citation in text: [1] -
Lu, H.; Hu, Y.; Jiang, H.; Wojtas, L.; Zhang, X. P. Org. Lett. 2012, 14, 5158–5161. doi:10.1021/ol302511f
Return to citation in text: [1] -
Horino, Y.; Yamamoto, T.; Ueda, K.; Kuroda, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2809–2811. doi:10.1021/ja808780r
Return to citation in text: [1] -
Bhunia, S.; Ghorpade, S.; Huple, D. B.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 2939–2942. doi:10.1002/anie.201108027
Return to citation in text: [1] -
DeBoef, B.; Pastine, S. J.; Sames, D. J. Am. Chem. Soc. 2004, 126, 6556–6557. doi:10.1021/ja049111e
Return to citation in text: [1] -
Pan, S.; Endo, K.; Shibata, T. Org. Lett. 2011, 13, 4692–4695. doi:10.1021/ol201907w
Return to citation in text: [1] -
Obora, Y.; Ogawa, S.; Yamamoto, N. J. Org. Chem. 2012, 77, 9429–9433. doi:10.1021/jo3019347
Return to citation in text: [1] -
Wang, Z.; Zhang, Y.; Fu, H.; Jiang, Y.; Zhao, Y. Org. Lett. 2008, 10, 1863–1866. doi:10.1021/ol800593p
Return to citation in text: [1] -
Zhang, S.-Y.; Tu, Y.-Q.; Fan, C.-A.; Zhang, F.-M.; Shi, L. Angew. Chem., Int. Ed. 2009, 48, 8761–8765. doi:10.1002/anie.200903960
Return to citation in text: [1] -
Pan, S.; Liu, J.; Li, H.; Wang, Z.; Guo, X.; Li, Z. Org. Lett. 2010, 12, 1932–1935. doi:10.1021/ol100670m
Return to citation in text: [1] -
Bloom, S.; Pitts, C. R.; Woltornist, R.; Griswold, A.; Holl, M. G.; Lectka, T. Org. Lett. 2013, 15, 1722–1724. doi:10.1021/ol400424s
Return to citation in text: [1] -
Vadola, P. A.; Sames, D. J. Am. Chem. Soc. 2009, 131, 16525–16528. doi:10.1021/ja906480w
Return to citation in text: [1] -
Yoshikai, N.; Mieczkowski, A.; Matsumoto, A.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2010, 132, 5568–5569. doi:10.1021/ja100651t
Return to citation in text: [1] -
Qian, B.; Xie, P.; Xie, Y.; Huang, H. Org. Lett. 2011, 13, 2580–2583. doi:10.1021/ol200684b
Return to citation in text: [1] -
Liu, X.; Sun, B.; Xie, Z.; Qin, X.; Liu, L.; Lou, H. J. Org. Chem. 2013, 78, 3104–3112. doi:10.1021/jo4000674
Return to citation in text: [1] -
Chen, L.; Shi, E.; Liu, Z.; Chen, S.; Wei, W.; Li, H.; Xu, K.; Wan, X. Chem.–Eur. J. 2011, 17, 4085–4089. doi:10.1002/chem.201100192
Return to citation in text: [1] -
He, T.; Yu, L.; Zhang, L.; Wang, L.; Wang, M. Org. Lett. 2011, 13, 5016–5019. doi:10.1021/ol201779n
Return to citation in text: [1] -
Huang, J.; Li, L.-T.; Li, H.-Y.; Husan, E.; Wang, P.; Wang, B. Chem. Commun. 2012, 48, 10204–10206. doi:10.1039/c2cc35450k
Return to citation in text: [1] -
Li, L.-T.; Li, H.-Y.; Xing, L.-J.; Wen, L.-J.; Wang, P.; Wang, B. Org. Biomol. Chem. 2012, 10, 9519–9522. doi:10.1039/c2ob26636a
Return to citation in text: [1] -
Mai, W.-P.; Wang, H.-H.; Li, Z.-C.; Yuan, J.-W.; Xiao, Y.-M.; Yang, L.-R.; Mao, P.; Qu, L.-B. Chem. Commun. 2012, 48, 10117–10119. doi:10.1039/c2cc35279f
Return to citation in text: [1] -
Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287–1292. doi:10.1002/adsc.201100920
Return to citation in text: [1] -
Shi, E.; Shao, Y.; Chen, S.; Hu, H.; Liu, Z.; Zhang, J.; Wan, X. Org. Lett. 2012, 14, 3384–3387. doi:10.1021/ol3013606
Return to citation in text: [1] -
Wang, F.-F.; Luo, C.-P.; Wang, Y.; Deng, G.; Yang, L. Org. Biomol. Chem. 2012, 10, 8605–8608. doi:10.1039/c2ob26604k
Return to citation in text: [1] -
Zhu, Y.-p.; Jia, F.-c.; Liu, M.-c.; Wu, L.-m.; Cai, Q.; Gao, Y.; Wu, A.-x. Org. Lett. 2012, 14, 5378–5381. doi:10.1021/ol302613q
Return to citation in text: [1] -
Zhu, Y.-p.; Liu, M.-c.; Jia, F.-c.; Yuan, J.-j.; Gao, Q.-h.; Lian, M.; Wu, A.-x. Org. Lett. 2012, 14, 3392–3395. doi:10.1021/ol301366p
Return to citation in text: [1] -
Xiang, J.; Fuchs, P. L. J. Am. Chem. Soc. 1996, 118, 11986–11987. doi:10.1021/ja962790b
Return to citation in text: [1] -
Jang, Y.-J.; Shih, Y.-K.; Liu, J.-Y.; Kuo, W.-Y.; Yao, C.-F. Chem.–Eur. J. 2003, 9, 2123–2128. doi:10.1002/chem.200204571
Return to citation in text: [1] -
Cui, Z.; Shang, X.; Shao, X.-F.; Liu, Z.-Q. Chem. Sci. 2012, 3, 2853–2858. doi:10.1039/c2sc20712e
Return to citation in text: [1] -
Forgione, P.; Brochu, M.-C.; St-Onge, M.; Thesen, K. H.; Bailey, M. D.; Bilodeau, F. J. Am. Chem. Soc. 2006, 128, 11350–11351. doi:10.1021/ja063511f
Return to citation in text: [1] -
Gooßen, L. J.; Rudolphi, F.; Oppel, C.; Rodríguez, N. Angew. Chem., Int. Ed. 2008, 47, 3043–3045. doi:10.1002/anie.200705127
Return to citation in text: [1] -
Kim, H.; Lee, P. H. Adv. Synth. Catal. 2009, 351, 2827–2832. doi:10.1002/adsc.200900502
Return to citation in text: [1] -
Wang, Z.; Ding, Q.; He, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 863–865. doi:10.1039/b821870f
Return to citation in text: [1] -
Zhang, F.; Greaney, M. F. Angew. Chem., Int. Ed. 2010, 49, 2768–2771. doi:10.1002/anie.200906921
Return to citation in text: [1] -
Wang, Z. T.; Zhu, L.; Yin, F.; Su, Z. Q.; Li, Z.; Li, C. J. Am. Chem. Soc. 2012, 134, 4258–4263. doi:10.1021/ja210361z
Return to citation in text: [1] -
Yin, F.; Wang, Z.; Li, Z.; Li, C. J. Am. Chem. Soc. 2012, 134, 10401–10404. doi:10.1021/ja3048255
Return to citation in text: [1] -
Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s
Return to citation in text: [1] -
Liu, Z.-Q.; Sun, L.; Wang, J.-G.; Han, J.; Zhao, Y.-K.; Zhou, B. Org. Lett. 2009, 11, 1437–1439. doi:10.1021/ol900145u
Return to citation in text: [1] -
Yang, H.; Yan, H.; Sun, P.; Zhu, Y.; Lu, L.; Liu, D.; Rong, G.; Mao, J. Green Chem. 2013, 15, 976–981. doi:10.1039/c3gc37131j
Return to citation in text: [1] -
Chowdhury, S.; Roy, S. J. Org. Chem. 1997, 62, 199–200. doi:10.1021/jo951991f
Return to citation in text: [1]
| 69. | Liu, Z.-Q.; Sun, L.; Wang, J.-G.; Han, J.; Zhao, Y.-K.; Zhou, B. Org. Lett. 2009, 11, 1437–1439. doi:10.1021/ol900145u |
| 70. | Yang, H.; Yan, H.; Sun, P.; Zhu, Y.; Lu, L.; Liu, D.; Rong, G.; Mao, J. Green Chem. 2013, 15, 976–981. doi:10.1039/c3gc37131j |
| 71. | Chowdhury, S.; Roy, S. J. Org. Chem. 1997, 62, 199–200. doi:10.1021/jo951991f |
| 1. | Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094–5115. doi:10.1002/anie.200806273 |
| 2. | Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074–1086. doi:10.1021/ar9000058 |
| 3. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n |
| 4. | Coperet, C. Chem. Rev. 2010, 110, 656–680. doi:10.1021/cr900122p |
| 5. | Werner, H. Angew. Chem., Int. Ed. 2010, 49, 4714–4728. doi:10.1002/anie.201000306 |
| 6. | Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293–1314. doi:10.1021/cr100198w |
| 7. | Campbell, A. N.; Stahl, S. S. Acc. Chem. Res. 2012, 45, 851–863. doi:10.1021/ar2002045 |
| 8. | Kozhushkov, S. I.; Ackermann, L. Chem. Sci. 2013, 4, 886–896. doi:10.1039/c2sc21524a |
| 24. | Pastine, S. J.; Gribkov, D. V.; Sames, D. J. Am. Chem. Soc. 2006, 128, 14220–14221. doi:10.1021/ja064481j |
| 25. | Rankin, M. A.; Schatte, G.; McDonald, R.; Stradiotto, M. J. Am. Chem. Soc. 2007, 129, 6390–6391. doi:10.1021/ja071684e |
| 26. | Deng, G.; Zhao, L.; Li, C.-J. Angew. Chem., Int. Ed. 2008, 47, 6278–6282. doi:10.1002/anie.200801544 |
| 27. | Wang, M.-Z.; Zhou, C.-Y.; Wong, M.-K.; Che, C.-M. Chem.–Eur. J. 2010, 16, 5723–5735. doi:10.1002/chem.200902387 |
| 60. | Cui, Z.; Shang, X.; Shao, X.-F.; Liu, Z.-Q. Chem. Sci. 2012, 3, 2853–2858. doi:10.1039/c2sc20712e |
| 19. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 20. | Xie, J.; Huang, Z.-Z. Angew. Chem., Int. Ed. 2010, 49, 10181–10185. doi:10.1002/anie.201004940 |
| 21. | Yang, F.; Li, J.; Xie, J.; Huang, Z.-Z. Org. Lett. 2010, 12, 5214–5217. doi:10.1021/ol102252n |
| 22. | Rout, S. K.; Guin, S.; Ghara, K. K.; Banerjee, A.; Patel, B. K. Org. Lett. 2012, 14, 3982–3985. doi:10.1021/ol301756y |
| 23. | Xia, R.; Niu, H.-Y.; Qu, G.-R.; Guo, H.-M. Org. Lett. 2012, 14, 5546–5549. doi:10.1021/ol302640e |
| 61. | Forgione, P.; Brochu, M.-C.; St-Onge, M.; Thesen, K. H.; Bailey, M. D.; Bilodeau, F. J. Am. Chem. Soc. 2006, 128, 11350–11351. doi:10.1021/ja063511f |
| 62. | Gooßen, L. J.; Rudolphi, F.; Oppel, C.; Rodríguez, N. Angew. Chem., Int. Ed. 2008, 47, 3043–3045. doi:10.1002/anie.200705127 |
| 63. | Kim, H.; Lee, P. H. Adv. Synth. Catal. 2009, 351, 2827–2832. doi:10.1002/adsc.200900502 |
| 64. | Wang, Z.; Ding, Q.; He, X.; Wu, J. Org. Biomol. Chem. 2009, 7, 863–865. doi:10.1039/b821870f |
| 65. | Zhang, F.; Greaney, M. F. Angew. Chem., Int. Ed. 2010, 49, 2768–2771. doi:10.1002/anie.200906921 |
| 66. | Wang, Z. T.; Zhu, L.; Yin, F.; Su, Z. Q.; Li, Z.; Li, C. J. Am. Chem. Soc. 2012, 134, 4258–4263. doi:10.1021/ja210361z |
| 67. | Yin, F.; Wang, Z.; Li, Z.; Li, C. J. Am. Chem. Soc. 2012, 134, 10401–10404. doi:10.1021/ja3048255 |
| 68. | Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s |
| 12. | Guin, S.; Rout, S. K.; Banerjee, A.; Nandi, S.; Patel, B. K. Org. Lett. 2012, 14, 5294–5297. doi:10.1021/ol302438z |
| 13. | Rousseaux, S.; Liégault, B.; Fagnou, K. Chem. Sci. 2012, 3, 244–248. doi:10.1039/c1sc00458a |
| 14. | Solé, D.; Mariani, F.; Fernández, I.; Sierra, M. A. J. Org. Chem. 2012, 77, 10272–10284. doi:10.1021/jo301924e |
| 15. | Wasa, M.; Chan, K. S. L.; Zhang, X.-G.; He, J.; Miura, M.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 18570–18572. doi:10.1021/ja309325e |
| 16. | Yin, Z.; Sun, P. J. Org. Chem. 2012, 77, 11339–11344. doi:10.1021/jo302125h |
| 17. | Saget, T.; Perez, D.; Cramer, N. Org. Lett. 2013, 15, 1354–1357. doi:10.1021/ol400380y |
| 18. | Zhang, S.-Y.; He, G.; Nack, W. A.; Zhao, Y.; Li, Q.; Chen, G. J. Am. Chem. Soc. 2013, 135, 2124–2127. doi:10.1021/ja312277g |
| 58. | Xiang, J.; Fuchs, P. L. J. Am. Chem. Soc. 1996, 118, 11986–11987. doi:10.1021/ja962790b |
| 9. | Chatani, N.; Asaumi, T.; Yorimitsu, S.; Ikeda, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 2001, 123, 10935–10941. doi:10.1021/ja011540e |
| 10. | Jiang, X. F.; Shen, M.; Tang, Y.; Li, C. Tetrahedron Lett. 2005, 46, 487–489. doi:10.1016/j.tetlet.2004.11.113 |
| 11. | Song, C.-X.; Cai, G.-X.; Farrell, T. R.; Jiang, Z.-P.; Li, H.; Gan, L.-B.; Shi, Z.-J. Chem. Commun. 2009, 6002–6004. doi:10.1039/b911031c |
| 59. | Jang, Y.-J.; Shih, Y.-K.; Liu, J.-Y.; Kuo, W.-Y.; Yao, C.-F. Chem.–Eur. J. 2003, 9, 2123–2128. doi:10.1002/chem.200204571 |
| 37. | DeBoef, B.; Pastine, S. J.; Sames, D. J. Am. Chem. Soc. 2004, 126, 6556–6557. doi:10.1021/ja049111e |
| 38. | Pan, S.; Endo, K.; Shibata, T. Org. Lett. 2011, 13, 4692–4695. doi:10.1021/ol201907w |
| 39. | Obora, Y.; Ogawa, S.; Yamamoto, N. J. Org. Chem. 2012, 77, 9429–9433. doi:10.1021/jo3019347 |
| 44. | Vadola, P. A.; Sames, D. J. Am. Chem. Soc. 2009, 131, 16525–16528. doi:10.1021/ja906480w |
| 45. | Yoshikai, N.; Mieczkowski, A.; Matsumoto, A.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2010, 132, 5568–5569. doi:10.1021/ja100651t |
| 46. | Qian, B.; Xie, P.; Xie, Y.; Huang, H. Org. Lett. 2011, 13, 2580–2583. doi:10.1021/ol200684b |
| 47. | Liu, X.; Sun, B.; Xie, Z.; Qin, X.; Liu, L.; Lou, H. J. Org. Chem. 2013, 78, 3104–3112. doi:10.1021/jo4000674 |
| 35. | Horino, Y.; Yamamoto, T.; Ueda, K.; Kuroda, S.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 2809–2811. doi:10.1021/ja808780r |
| 36. | Bhunia, S.; Ghorpade, S.; Huple, D. B.; Liu, R.-S. Angew. Chem., Int. Ed. 2012, 51, 2939–2942. doi:10.1002/anie.201108027 |
| 48. | Chen, L.; Shi, E.; Liu, Z.; Chen, S.; Wei, W.; Li, H.; Xu, K.; Wan, X. Chem.–Eur. J. 2011, 17, 4085–4089. doi:10.1002/chem.201100192 |
| 49. | He, T.; Yu, L.; Zhang, L.; Wang, L.; Wang, M. Org. Lett. 2011, 13, 5016–5019. doi:10.1021/ol201779n |
| 50. | Huang, J.; Li, L.-T.; Li, H.-Y.; Husan, E.; Wang, P.; Wang, B. Chem. Commun. 2012, 48, 10204–10206. doi:10.1039/c2cc35450k |
| 51. | Li, L.-T.; Li, H.-Y.; Xing, L.-J.; Wen, L.-J.; Wang, P.; Wang, B. Org. Biomol. Chem. 2012, 10, 9519–9522. doi:10.1039/c2ob26636a |
| 52. | Mai, W.-P.; Wang, H.-H.; Li, Z.-C.; Yuan, J.-W.; Xiao, Y.-M.; Yang, L.-R.; Mao, P.; Qu, L.-B. Chem. Commun. 2012, 48, 10117–10119. doi:10.1039/c2cc35279f |
| 53. | Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287–1292. doi:10.1002/adsc.201100920 |
| 54. | Shi, E.; Shao, Y.; Chen, S.; Hu, H.; Liu, Z.; Zhang, J.; Wan, X. Org. Lett. 2012, 14, 3384–3387. doi:10.1021/ol3013606 |
| 55. | Wang, F.-F.; Luo, C.-P.; Wang, Y.; Deng, G.; Yang, L. Org. Biomol. Chem. 2012, 10, 8605–8608. doi:10.1039/c2ob26604k |
| 56. | Zhu, Y.-p.; Jia, F.-c.; Liu, M.-c.; Wu, L.-m.; Cai, Q.; Gao, Y.; Wu, A.-x. Org. Lett. 2012, 14, 5378–5381. doi:10.1021/ol302613q |
| 57. | Zhu, Y.-p.; Liu, M.-c.; Jia, F.-c.; Yuan, J.-j.; Gao, Q.-h.; Lian, M.; Wu, A.-x. Org. Lett. 2012, 14, 3392–3395. doi:10.1021/ol301366p |
| 32. | Harden, J. D.; Ruppel, J. V.; Gao, G.-Y.; Zhang, X. P. Chem. Commun. 2007, 4644–4646. doi:10.1039/b710677g |
| 33. | Hung-Low, F.; Krogman, J. P.; Tye, J. W.; Bradley, C. A. Chem. Commun. 2012, 48, 368–370. doi:10.1039/c1cc15458c |
| 34. | Lu, H.; Hu, Y.; Jiang, H.; Wojtas, L.; Zhang, X. P. Org. Lett. 2012, 14, 5158–5161. doi:10.1021/ol302511f |
| 28. | Shi, L.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M.; Fan, C.-A.; Zhao, Y.-M.; Xia, W.-J. J. Am. Chem. Soc. 2005, 127, 10836–10837. doi:10.1021/ja0528331 |
| 29. | Jo, E.-A.; Lee, J.-H.; Jun, C.-H. Chem. Commun. 2008, 5779–5781. doi:10.1039/b814166e |
| 30. | Rakshit, S.; Patureau, F. W.; Glorius, F. J. Am. Chem. Soc. 2010, 132, 9585–9587. doi:10.1021/ja104305s |
| 31. | Kuninobu, Y.; Nakahara, T.; Takeshima, H.; Takai, K. Org. Lett. 2013, 15, 426–428. doi:10.1021/ol303353m |
| 40. | Wang, Z.; Zhang, Y.; Fu, H.; Jiang, Y.; Zhao, Y. Org. Lett. 2008, 10, 1863–1866. doi:10.1021/ol800593p |
| 41. | Zhang, S.-Y.; Tu, Y.-Q.; Fan, C.-A.; Zhang, F.-M.; Shi, L. Angew. Chem., Int. Ed. 2009, 48, 8761–8765. doi:10.1002/anie.200903960 |
| 42. | Pan, S.; Liu, J.; Li, H.; Wang, Z.; Guo, X.; Li, Z. Org. Lett. 2010, 12, 1932–1935. doi:10.1021/ol100670m |
| 43. | Bloom, S.; Pitts, C. R.; Woltornist, R.; Griswold, A.; Holl, M. G.; Lectka, T. Org. Lett. 2013, 15, 1722–1724. doi:10.1021/ol400424s |
© 2013 Zhao et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)