Abstract
Sterically crowded diimine ligands with five aryl rings were prepared in one step in good yields using a micro-flow technique. X-ray crystallographic analysis revealed the detailed structure of the bulky ligands. The nickel complexes prepared from the ligands exerted high polymerization activity in the ethylene homopolymerization and copolymerization of ethylene with polar monomers.

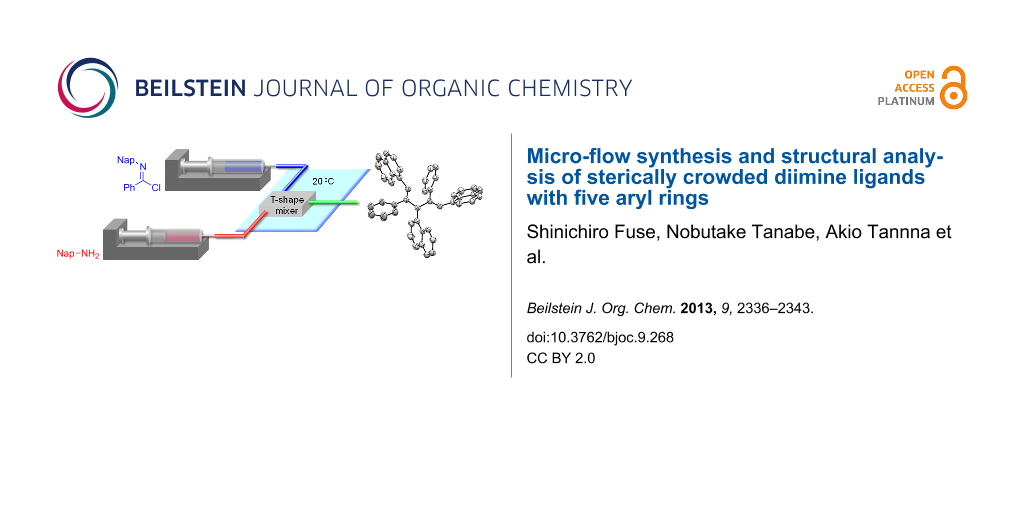
Graphical Abstract
Introduction
The design of a ligand is a key step in the development of new catalysts because the ligand framework influences the reactivity of the metal center. That is why sterically crowded and neutral-chelating diimine ligands have garnered a great deal of attention [1-17]. In recent years, N-aryl 1,3,5-triazapenta-1,4-dienes 1 and 2 have been reported, and they are useful with late transition metal olefin-polymerization catalysts [18,19], and for the stabilization and isolation of reactive metal species [20,21]. In 1997, Murillo and coworkers reported the synthesis of a neutral, and bulky chelating ligand 1, and its use in the formation of a Co complex (Figure 1) [22]. Stephan and coworkers reported the synthesis of a bulkier chelating ligand 2a, and its use in the formation of various metal complexes [20,21]. Rojas and coworkers reported the synthesis of a series of bulky chelating ligands 2b–g, and detailed their use in the preparation of ethylene polymerization catalysts [18,19]. We became curious about the structure and function of 1,2,3,4,5-pentaaryl-1,3,5-triazapenta-1,4-diene ligand 3, which is sterically more hindered, because there are as many as five aryl rings that can provide further opportunities to change and tune the steric and electrical environments of the ligands [23,24]. However, as far as we could ascertain, this has been reported only once [25]. In this pioneering work, 1,2,3,4,5-pentaphenyl-1,3,5-triazapenta-1,4-diene ligand 3a was prepared, but the report included neither the complexation nor a detailed structural study.
![[1860-5397-9-268-1]](/bjoc/content/figures/1860-5397-9-268-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Sterically crowded, and neutral chelating diimine ligands.
Figure 1: Sterically crowded, and neutral chelating diimine ligands.
Herein, we report an efficient micro-flow synthesis and structural analysis of sterically crowded 1,2,3,4,5-pentaaryl-1,3,5-triazapenta-1,4-diene ligands 3b and 3c, and their use in the copolymerization of ethylene and polar monomers. According to the previous report, 1,2,3,4,5-pentaphenyl-1,3,5-triazapenta-1,4-diene ligand 3a was prepared by the reaction of N,N′-diphenyl benzamidine and N-phenylbenzimidochloride in benzene in 13 days [25]. In the present study, we intended to prepare these ligands from readily available materials in only one step [18,21].
Results and Discussion
Two equivalents of N-naphthylbenzimidochloride (4) was reacted with one equivalent of naphthylamine (5) in CH2Cl2 in the presence of DIEA (N,N-diisopropylethylamine) at room temperature (Scheme 1). The reaction proceeded smoothly, and consumption of naphthylamine was confirmed by TLC analysis within 10 min. After an aqueous workup, the desired product 3b was obtained in a moderate yield (44%) with the concomitant generation of naphthylamine (5) and amide 8. Reportedly, 3a can form an HCl adduct, and the adduct decomposes to the corresponding amidine and imidochloride [25]. It is conceivable that 3b overreacted with DIEA·HCl to afford 6, or 4 and 7 in the reaction mixture and that 5 and 8 were generated from the hydrolysis of these compounds under aqueous workup conditions. We decided to use a micro-flow reactor in order to suppress the overreaction [26,27] because the micro-flow technique [28-35] enables the precise control of reaction time and temperature. The micro-flow system was made from simple and inexpensive laboratory instruments (syringes, syringe pumps, water bath, T-shape mixer, standard tubing and fittings), as shown in Figure 2. The T-shape mixer was made of stainless steel and immersed in a water bath (20 °C). A solution of 4 (0.1 M) in CH2Cl2, a solution of aryl amine 5 or 9 (0.1 M), and DIEA (0.7 M) in CH2Cl2 were introduced using syringe pumps at the indicated flow rates. The reaction was quenched by pouring the mixture into a saturated aqueous solution of NH4Cl in CH2Cl2. After an aqueous workup, the products 3b and 3c were purified by silica gel chromatography. Reaction time was controlled by changing the flow rates.
![[1860-5397-9-268-i1]](/bjoc/content/inline/1860-5397-9-268-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-step synthesis of the ligand 3b, and a plausible decomposition pathway for 3b to naphthylamine (5) and amide 8.
Scheme 1: One-step synthesis of the ligand 3b, and a plausible decomposition pathway for 3b to naphthylamine (...
![[1860-5397-9-268-2]](/bjoc/content/figures/1860-5397-9-268-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Micro-flow synthesis of the ligands 3b and 3c.
Figure 2: Micro-flow synthesis of the ligands 3b and 3c.
As expected, the yield of 3b was improved by reducing the reaction time (Table 1, entries 1–4). The highest yield was observed for a reaction time of 38 s (Table 1, entry 4). A further shortening of the reaction time resulted in a reduction in the yield because of substrate recovery (Table 1, entry 5).
Table 1: Micro-flow synthesis of ligands 3b and 3c.
| entry |
flow rate A
[μL/min] |
flow rate B
[μL/min] |
time
[s] |
Ar–NH2 |
yielda
[%] |
|---|---|---|---|---|---|
| 1b | 54 | 27 | 300 | 5 | 65 |
| 2b | 106 | 53 | 150 | 5 | 72 |
| 3b | 214 | 107 | 75 | 5 | 75 |
| 4b | 426 | 213 | 38 | 5 | 84 |
| 5b | 854 | 427 | 19 | 5 | 66 |
| 6c | 214 | 107 | 50 | 9 | 55 |
| 7c | 426 | 213 | 25 | 9 | 69 |
| 8c | 854 | 427 | 13 | 9 | 66 |
aIsolated yield. bReaction tube volume is 400 μL. cReaction tube volume is 266 μL.
The structure of the ligand 3b was unambiguously determined by 1H NMR, 13C NMR, IR, HRMS and X-ray crystallographic analysis [36] (Figure 3). The ORTEP structure of 3b showed that in the solid state the ligand adopts a non-planar arrangement similar to the previously reported ligands 2a–c, and 2e–g [18,19,21]. In ligand 3b, N(1) and N(2) nearly occupied a common plane with C(1) and C(2), while N(3) was twisted out of this plane and was nearly perpendicular. The bond lengths for imines C(1)–N(1) and C(2)–N(3) were 1.273(4) and 1.278(4) Å, respectively, while the bond lengths for amines C(1)–N(2) and C(2)–N(2) were 1.420(4) and 1.421(4) Å, respectively. Reportedly, the two amine bond lengths were different in the case of ligands, 2a, 2b, 2e, 2f, and 2g for which N=C–N–C=N was not in a common plane. On the other hand, the two amine bond lengths were nearly identical in the case of ligand 2c where N=C–N–C=N was in a common plane. Interestingly, in the case of ligand 3b, the bond lengths of the two amines, C(1)–N(2) and C(2)–N(2) were nearly identical, although N=C–N–C=N was not in a common plane.
![[1860-5397-9-268-3]](/bjoc/content/figures/1860-5397-9-268-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP drawing of 3b. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] are as follows: C(1)–N(1) 1.273(4), C(1)–N(2) 1.420(4), C(2)–N(2) 1.421(4), C(2)–N(3) 1.278(4), N(1)–C(1)–N(2) 115.3(3), C(1)–N(2)–C(2) 117.0(3), N(2)–C(2)–N(3) 117.8(3).
Figure 3: ORTEP drawing of 3b. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms are omitte...
The asymmetric ligand 3c was obtained by the coupling of N-naphthylbenzimidochloride (4) with aniline (9). The product was obtained in a satisfactory yield (69%) under the conditions of entry 7 (25 s), as shown in Table 1. We speculated that the slightly lower yield of 3c compared to 3b came from the instability of 3c during purification process. The compound 3c was less stable than 3b. Rojas et al. reported that the regioselectivity in the nucleophilic addition of an amidine to an imidochloride depends on the employed reaction conditions, – in particular, the order of addition and the base selection [37] – and the symmetric ligands were obtained through the coupling of N-2,6-di(iPr)-phenylbenzimidochloride with aryl amines in the presence of Et3N in toluene [18,19]. Interestingly, in our case, only the asymmetric ligand 3c was obtained, although similar reaction conditions were employed (Scheme 2). The result showed that the nucleophilic addition of amidine 10 occurred from the sterically more hindered nitrogen atom (path b).
![[1860-5397-9-268-i2]](/bjoc/content/inline/1860-5397-9-268-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the ligand 3c through the coupling of 4 and 9.
Scheme 2: Synthesis of the ligand 3c through the coupling of 4 and 9.
The structure of the asymmetric ligand 3c was unambiguously determined by 1H NMR, IR, HRMS and X-ray crystallographic analysis (Figure 4). The ORTEP structure of 3c showed that in the solid state, the ligand adopted a non-planar arrangement similar to that of 3b. In the ligand 3c, N(1) and N(2) occupied a plane that was near that of C(1) and C(2), while N(3) was twisted out of this plane and was almost perpendicular. The bond lengths for imines C(1)–N(1) and C(2)–N(3) were 1.269(3) and 1.274(3) Å, respectively, while the bond lengths for amines C(1)–N(2) and C(2)–N(2) were 1.413(3) and 1.411(3) Å, respectively. The bond lengths for amines C(1)–N(2) and C(2)–N(2) were nearly identical, although N=C–N–C=N was not in a common plane. These features were similar to that of 3b.
![[1860-5397-9-268-4]](/bjoc/content/figures/1860-5397-9-268-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawing of 3c. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°] are as follows: C(1)–N(1) 1.269(3), C(1)–N(2) 1.413(3), C(2)–N(2) 1.411(3), C(2)–N(3) 1.274(3), N(1)–C(1)–N(2) 116.5(2), C(1)–N(2)–C(2) 119.1(2), N(2)–C(2)–N(3) 117.4(2).
Figure 4: ORTEP drawing of 3c. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms are omitte...
A complexation of the synthesized ligands 3b and 3c with nickel(II) was performed (Scheme 3) in accordance with a reported procedure [38]. An equimolar amount of NiBr2(dme) (dme = 1,2-dimethoxyethane) and the synthesized ligands were mixed in CH2Cl2 and stirred for 4 h at room temperature. Free ligands 3b or 3c were not observed by 1H NMR analysis of the obtained crude mixtures. NMR characterization of the complexes 12 and 13 was poor due to the paramagnetic nature of the pseudo-tetrahedral nickel centers. In the case of 12, green needle-like crystals suitable for X-ray crystallographic analysis were obtained (Figure 5). The molecular structure confirmed the formation of a six-membered chelate ring by the ligand in a N,N binding mode to the nickel dibromide. All the aryl rings were almost perpendicular to the chelate plane probably due to the strong steric repulsions among the aryl rings. The tetrahedral geometry around nickel was distorted similar to the previously reported nickel complexes of 2b, 2f and 2g [18,19]. For example, Br(1) was almost perpendicular to the plane of the metal-containing ring. This can be observed in the corresponding angle N(1)–Ni(1)–Br(1) 102.2(3)°, while the angle of N(1)–Ni(1)–Br(2) was 120.9(3)°. The C(1)–N(1), C(2)–N(3), C(1)–N(2) and C(2)–N(2) bond lengths were 1.277(11) Å, 1.305(11) Å, 1.393(11) Å and 1.396(11) Å respectively, indicating the double and single bond character around the imines and amine nitrogen, respectively.
![[1860-5397-9-268-i3]](/bjoc/content/inline/1860-5397-9-268-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Complexation of the ligands 3b and 3c with NiBr2(dme).
Scheme 3: Complexation of the ligands 3b and 3c with NiBr2(dme).
![[1860-5397-9-268-5]](/bjoc/content/figures/1860-5397-9-268-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP drawing of 12. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms and cocrystallized CH2Cl2 are omitted for clarity. Selected bond lengths [Å] and angles [°] are as follows: C(1)–N(1) 1.277(11), C(1)–N(2) 1.393(11), C(2)–N(2) 1.396(11), C(2)–N(3) 1.305(11), Ni(1)–N(1) 1.947(8), Ni(1)–N(3) 1.943(8), N(1)–C(1)–N(2) 122.8(8), C(1)–N(2)–C(2) 126.2(7), N(2)–C(2)–N(3) 121.2(8), C(2)–N(3)–Ni(1) 127.5(6), N(1)–Ni(1)–N(3) 89.3(3), C(1)–N(1)–Ni(1), 127.7(6), N(1)–Ni(1)–Br(1) 102.2(3), N(1)–Ni(1)–Br(2) 120.9(3).
Figure 5: ORTEP drawing of 12. Thermal ellipsoids are set at 35% probability level. Hydrogen atoms and cocrys...
A series of olefin polymerizations were briefly tested as shown in Table 2. Both catalysts 12 and 13 exerted high activity for the ethylene homopolymerization (Table 2, entries 1 and 2). As far as we could ascertain, neither propene homopolymerization nor copolymerization of ethylene with polar monomers by using the catalysts derived from N-aryl-1,3,5-triazapenta-1,4-dienes 1–3 have been reported [18,19]. Thus, we tested the polymerization activity of the catalyst 12 in the copolymerization of ethylene with 5-norbornen-2-ol (NBO) and ethyl acrylate (EtA) (Table 2, entries 3 and 4) and propene homopolymerization (Table 2, entry 5). Although the propene homopolymerization activity of catalyst 12 was moderate, catalyst 12 exerted a high activity in the copolymerization of ethylene with both polar monomers.
Table 2: Evaluation of polymerization activities of nickel complexes 12 and 13.a
| entry | catalystb | monomerc |
temperature, time
[°C, h] |
Vp
[kg/mol·h] |
|---|---|---|---|---|
| 1 | 12 | ethylene | 60, 0.5 | 140 |
| 2 | 13 | ethylene | 60, 0.5 | 110 |
| 3 | 12 | ethylene/NBO | 60, 1 | 25 |
| 4 | 12 | ethylene/EtA | 60, 1 | 35 |
| 5 | 12 | propene | 60, 1 | 5 |
aEntries 1–4 were carried out in a 2 L autoclave reactor in 1 L toluene in the presence of 500 equiv of modified methyl aluminoxane (MMAO)[39] as cocatalyst and 400 psig of ethylene. Entry 5 was carried out in a 2 L autoclave reactor in 750 mL propene in the presence of 500 equiv of MMAO as the cocatalyst. bEntries 1, 4 and 5 were carried out using 0.11 mmol of the catalyst. Entries 2 and 3 were carried out using 0.10 and 0.09 mmol of the catalysts, respectively. cEntries 3 and 4 were carried out using 500 equiv of NBO or EtA.
Conclusion
In summary, sterically crowded diimine ligands 3b and 3c were prepared in one step in good yields using a micro-flow technique. One of the advantages of using microreactors is the ease of scale-up. It should be possible to scale-up our developed process by either continuous running or by increasing the number of the microreactors. X-ray crystallographic analysis revealed the detailed structure of ligands 3b and 3c. Interestingly, 3c retained an asymmetric structure which ran contrary to a previous report. Unexpectedly, both bond lengths of the two amines C(1)–N(2) and C(2)–N(2) in both ligands were nearly identical, although N=C–N–C=N was not in a common plane. The complexation of 3b and 3c with nickel afforded 12 and 13. X-ray crystallographic analysis of 12 revealed that all the aryl rings are nearly perpendicular to the chelate plane. The nickel complex 12 exerted a high polymerization activity in both ethylene homopolymerization and the copolymerization of ethylene with polar monomers. The synthesized ligands 3b and 3c retained as many as five aryl rings, which offered another opportunity to change the steric and electric environment. The developed process should be valuable for the preparation of various 1,2,3,4,5-pentaaryl-1,3,5-triazapenta-1,4-diene ligands and for the creation of novel and useful catalysts.
Experimental
General
NMR spectra were recorded on a JEOL Model ECP-400 (400 MHz for 1H, 100 MHz for 13C) instrument in the indicated solvent. Chemical shifts are reported in units of parts per million (ppm) relative to the signal (0.00 ppm) for internal tetramethylsilane for solutions in CDCl3 (7.26 ppm for 1H, 77.0 ppm for 13C). Multiplicities are reported by using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; and, J, coupling constants in Hertz. IR spectra were recorded on a Perkin-Elmer Spectrum One FTIR spectrometer. HRMS (ESI–TOF) was measured with a Waters LCT PremierTM XE. All reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60F-254) with UV light, visualized by ceric sulfate solution. Flash column chromatography was performed on Silica Gel 60 N, purchased from Kanto Chemical Co. The T-shape mixer (Flom Co. Ltd., #9513, 19 mm × 28 mm × 8 mm, diameter 0.6 mm) was made of stainless steel and had a T-shape channel. The reaction tube (diameter 0.5 mm) was made of Teflon®. A Harvard Pump 11 Plus Single Syringe (HARVARD apparatus), a KDS 100 syringe pump, and a KDS 200 syringe pump (KD Scientific) were used to inject compounds into the T-shape mixers. The workup process included quenching of the reactions, liquid–liquid extraction, washing and drying, and was performed using a Zodiac CCX-1200 (Tokyo Rikakikai Co., Ltd.). Chromatographic separation was performed using a Purif®-α2 (Shoko Scientific Co., Ltd.).
Experimental details
General procedure for the preparation of imidochlorides: The mixture of N-(1-naphthyl)benzamide and SOCl2 (2 mL/mmol amide) was stirred at 65 °C for 4 h. The reaction mixture was concentrated in vacuo. The residue was used for the next reaction without further purification.
General procedure for micro-flow synthesis of 1,2,3,4,5-pentaaryl-1,3,5-triazapenta-1,4-dienes 3b and 3c: A T-shape mixer and reaction tube were immersed in a water bath (20 °C). Syringe pumps and a mixer were connected using a Teflon® tube (diameter 0.25 mm). Imidochloride 4 was dried azeotropically with toluene. Aryl amine 5 or 9 (0.1 M), DIEA (0.7 M) and imidochloride 4 (0.1 M) were dissolved in CH2Cl2 under an argon atmosphere and were stored in syringes. Each solution was introduced to a T-shape mixer using the syringe pump. The mixed solution went through the reaction tube, and the resultant solution was poured into vigorously stirred saturated aqueous NH4Cl (1.5 mL) in CH2Cl2 (2 mL). After being stirred for several minutes, Et2O (30 mL) was added to the reaction mixture under vigorous stirring for 30 s. The aqueous layer was separated and added to saturated aqueous NaHCO3 (2 mL) after being vigorously stirred for 30 s. The aqueous layer was separated and brine (2 mL) was added followed by vigorous stirring for 30 s. After removing the aqueous layer, the organic layers were dried over Na2SO4 (10 g) and concentrated in vacuo. The residue was purified by column chromatography on silica gel (0% to 10% Et2O in hexane with 1% Et3N) to give 1,2,3,4,5-pentaaryl-1,3,5-triazapenta-1,4-diene 3b or 3c.
1,3,5-Trinaphthyl-2,4-diphenyl-triazapenta-1,4-diene (3b): 1H NMR (400 MHz, CDCl3) δ 8.22 (brd, J = 5.8 Hz, 1H), 8.15 (brs, 1H), 7.89 (brd, J = 5.4 Hz, 1H), 7.74–7.90 (m, 2H), 7.73 (brd, J = 6.8 Hz, 2H), 7.58 (brd, J = 7.8 Hz, 1H), 7.10–7.58 (m, 20H), 7.07 (t, J = 7.8 Hz, 1H), 7.39 (brd, J = 7.8, 1H), 6.64 (brs, 1H); 13C NMR (67.8 MHz, CDCl3) δ 156.2, 145.6, 136.1, 135.2, 134.7, 134.0, 130.1, 129.5, 128.8, 128.3, 128.2, 127.2, 126.3, 125.9, 125.8, 125.6, 125.2, 125.0, 123.6, 123.5, 123.0, 122.5, 121.6, 118.0, 115.7; FTIR (neat) 3055, 1624, 1572, 1529, 1495, 1393, 791, 771 cm−1; HRMS (ESI–TOF, m/z): [M + H]+ calcd. for C44H32N3, 602.2596; found, 602.2613.
1,3-Dinaphthyl-2,4,5-triphenyl-triazapenta-1,4-diene (3c): 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 6.4 Hz, 2H), 8.25 (d, J = 8.3 Hz, 2H), 7.78 (brd, J = 7.8 Hz, 1H), 7.73 (d, J = 7.8 Hz, 2H), 7.61 (d, J = 7.8 Hz, 1H), 7.36–7.60 (m, 9H), 7.40–7.20 (m, 2H), 7.16 (t, J = 7.8 Hz, 1H), 7.05–6.60 (m, 7H), 6.54 (brs, 1H), 5.71 (brd, J = 6.8, 1H); FTIR (neat) 3056, 1631, 1595, 1524, 1497, 1438, 1323, 775, 698 cm−1; HRMS (ESI–TOF, m/z): [M + H]+ calcd. for C40H30N3, 552.2440; found, 552.2435.
General procedure for the preparation of nickel complexes 12 and 13
The following manipulations were performed under an inert atmosphere using standard glove box techniques. A solution of the prepared ligand 3b or 3c (1 equiv) and NiBr2(dme) (1 equiv) in dry CH2Cl2 (50 mL/mmol) was stirred for 4 h at room temperature. The obtained crude mixture was used for the polymerization without purification.
Procedure for the preparation of nickel complex 12 crystals
The following manipulations were performed under an inert atmosphere using standard glove box techniques. A solution of the prepared ligand 3b (66 mg, 0.11 mmol) in 40 mL of dry CH2Cl2 was slowly added to NiBr2(dme) (34 mg, 0.11 mmol). The resultant mixture was stirred for 1 h at room temperature. Then, the stirring was stopped and the mixture was allowed to stand overnight at room temperature. Green colored needle-like crystals of 12, suitable for X-ray analysis were obtained.
Procedure for the homopolymerization of ethylene
To a 2 L autoclave reactor, 1,000 mL of dry toluene and MMAO (500 equiv, 6.5 wt % in toluene) were added. The resultant mixture was heated to 60 °C, then the crude nickel complex (1 equiv) was injected under an ethylene pressure of 400 psig, which was fed continuously at that pressure over the course of the reaction. After being stirred for 0.5 h, ethanol was added to quench the polymerization, and ethylene was vented. The resultant mixture was collected, and concentrated in vacuo to afford a crude polymer. Polymerization activities were calculated from the mass of the crude polymer that was obtained.
Procedure for the copolymerization of ethylene with polar monomers
To a 2 L autoclave reactor, 1,000 mL of dry toluene and comonomers (500 equiv) along with MMAO (500 equiv, 6.5 wt % in toluene) were added. The resultant mixture was heated to 60 °C, then the crude nickel complex (1 equiv) was injected under an ethylene pressure of 400 psig, which was fed continuously at that pressure over the course of the reaction. After being stirred for 1 h, ethanol was added to quench the polymerization, and ethylene was vented. The resultant mixture was collected, and concentrated in vacuo to afford the crude polymer. Polymerization activities were calculated from the mass of the crude polymer that was obtained.
Procedure for the homopolymerization of propene
To a 2 L autoclave reactor, MMAO (500 equiv, 6.5 wt % in toluene) and 750 mL of propene were added. Then the crude nickel complex (1 equiv) was injected with nitrogen. The resultant mixture was heated to 60 °C. After being stirred for 1 h, ethanol was added to quench the polymerization, and propene was vented. The resultant mixture was collected, and concentrated in vacuo to afford a crude polymer. Polymerization activities were calculated from the mass of the crude polymer that was obtained.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 214.4 KB | Download |
Acknowledgements
The authors thank Dr. Hidehiro Uekusa, Tokyo Institute of Technology for X-ray crystallographic analysis of intermediates and Dr. Shigekazu Ito, Tokyo Institute of Technology for fruitful discussion, and Global COE Program “Education and Research Center for Emergence of New Molecular Chemistry,” MEXT, Japan, for financial support.
References
-
Ittel, S. D.; Johnson, L. K.; Brookhart, M. Chem. Rev. 2000, 100, 1169. doi:10.1021/cr9804644
Return to citation in text: [1] -
Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471. doi:10.1021/cr030710y
Return to citation in text: [1] -
Dagorne, S.; Atwood, D. A. Chem. Rev. 2008, 108, 4037. doi:10.1021/cr078351k
Return to citation in text: [1] -
Nakamura, A.; Ito, S.; Nozaki, K. Chem. Rev. 2009, 109, 5215. doi:10.1021/cr900079r
Return to citation in text: [1] -
Chen, E. Y.-X. Chem. Rev. 2009, 109, 5157. doi:10.1021/cr9000258
Return to citation in text: [1] -
Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2009, 110, 890. doi:10.1021/cr900206p
Return to citation in text: [1] -
Driver, T. G. Org. Biomol. Chem. 2010, 8, 3831. doi:10.1039/c005219c
Return to citation in text: [1] -
Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Chem. Soc. Rev. 2010, 39, 712. doi:10.1039/B809912J
Return to citation in text: [1] -
Asay, M.; Jones, C.; Driess, M. Chem. Rev. 2010, 111, 354. doi:10.1021/cr100216y
Return to citation in text: [1] -
Nomura, K.; Zhang, S. Chem. Rev. 2010, 111, 2342. doi:10.1021/cr100207h
Return to citation in text: [1] -
Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824. doi:10.1021/cr9003242
Return to citation in text: [1] -
Delferro, M.; Marks, T. J. Chem. Rev. 2011, 111, 2450. doi:10.1021/cr1003634
Return to citation in text: [1] -
Partyka, D. V. Chem. Rev. 2011, 111, 1529. doi:10.1021/cr1002276
Return to citation in text: [1] -
Takeuchi, D. Macromol. Chem. Phys. 2011, 212, 1545. doi:10.1002/macp.201100182
Return to citation in text: [1] -
Gephart, R. T., III; Warren, T. H. Organometallics 2012, 31, 7728. doi:10.1021/om300840z
Return to citation in text: [1] -
Lang, H.; Jakob, A.; Milde, B. Organometallics 2012, 31, 7661. doi:10.1021/om300628g
Return to citation in text: [1] -
Li, T.; Schulz, S.; Roesky, P. W. Chem. Soc. Rev. 2012, 41, 3759. doi:10.1039/c2cs15343b
Return to citation in text: [1] -
Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Masuda, J. D.; Stephan, D. W. Can. J. Chem. 2005, 83, 477. doi:10.1139/v05-057
Return to citation in text: [1] [2] -
Masuda, J. D.; Stephan, D. W. Dalton Trans. 2006, 2089. doi:10.1039/B513531A
Return to citation in text: [1] [2] [3] [4] -
Cotton, F. A.; Daniels, L. M.; Matonic, J. H.; Wang, X. P.; Murillo, C. A. Polyhedron 1997, 16, 1177. doi:10.1016/s0277-5387(96)00366-x
Return to citation in text: [1] -
Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j
Return to citation in text: [1] -
Masui, H.; Fuse, S.; Takahashi, T. Org. Lett. 2012, 14, 4090. doi:10.1021/ol3017337
Return to citation in text: [1] -
Cooper, F. C.; Partridge, M. W.; Short, W. F. J. Chem. Soc. 1951, 391. doi:10.1039/jr9510000391
Return to citation in text: [1] [2] [3] -
Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.-i.; Fukase, K. Org. Lett. 2006, 9, 299. doi:10.1021/ol062777o
Return to citation in text: [1] -
Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2005, 127, 11666. doi:10.1021/ja0527424
Return to citation in text: [1] -
Luis, S. V.; Carcia-Verdugo, E., Eds. Chemical Reactions and Processes under Flow Conditions; Royal Society of Chemistry: Cambridge, 2010. doi:10.1039/9781847559739
Return to citation in text: [1] -
McMullen, J. P.; Jensen, K. F. Annu. Rev. Anal. Chem. 2010, 3, 19. doi:10.1146/annurev.anchem.111808.073718
Return to citation in text: [1] -
Fuse, S.; Tanabe, N.; Yoshida, M.; Yoshida, H.; Doi, T.; Takahashi, T. Chem. Commun. 2010, 46, 8722. doi:10.1039/c0cc02239j
Return to citation in text: [1] -
Suga, S.; Yamada, D.; Yoshida, J. Chem. Lett. 2010, 39, 404. doi:10.1246/cl.2010.404
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V. Mol. Diversity 2011, 15, 613. doi:10.1007/s11030-010-9282-1
Return to citation in text: [1] -
Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331. doi:10.1002/cssc.201000271
Return to citation in text: [1] -
Fuse, S.; Tanabe, N.; Takahashi, T. Chem. Commun. 2011, 47, 12661. doi:10.1039/c1cc15662d
Return to citation in text: [1] -
Fuse, S.; Mifune, Y.; Tanabe, N.; Takahashi, T. Org. Biomol. Chem. 2012, 10, 5205. doi:10.1039/c2ob25511a
Return to citation in text: [1] -
CCDC 934692 (3b), CCDC 934694 (3c), and 934693 (12) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Caris, R.; Peoples, B. C.; Valderrama, M.; Wu, G.; Rojas, R. J. Organomet. Chem. 2009, 694, 1795. doi:10.1016/j.jorganchem.2009.01.005
Return to citation in text: [1] -
Azoulay, J. D.; Rojas, R. S.; Serrano, A. V.; Ohtaki, H.; Galland, G. B.; Wu, G.; Bazan, G. C. Angew. Chem., Int. Ed. 2009, 48, 1089. doi:10.1002/anie.200804661
Return to citation in text: [1] -
MMAO is a modified methylaluminoxane activator containing 25% isobutyl aluminoxane prepared by the controlled hydrolysis of Me3Al and (iBu)3Al.
Return to citation in text: [1]
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 38. | Azoulay, J. D.; Rojas, R. S.; Serrano, A. V.; Ohtaki, H.; Galland, G. B.; Wu, G.; Bazan, G. C. Angew. Chem., Int. Ed. 2009, 48, 1089. doi:10.1002/anie.200804661 |
| 1. | Ittel, S. D.; Johnson, L. K.; Brookhart, M. Chem. Rev. 2000, 100, 1169. doi:10.1021/cr9804644 |
| 2. | Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471. doi:10.1021/cr030710y |
| 3. | Dagorne, S.; Atwood, D. A. Chem. Rev. 2008, 108, 4037. doi:10.1021/cr078351k |
| 4. | Nakamura, A.; Ito, S.; Nozaki, K. Chem. Rev. 2009, 109, 5215. doi:10.1021/cr900079r |
| 5. | Chen, E. Y.-X. Chem. Rev. 2009, 109, 5157. doi:10.1021/cr9000258 |
| 6. | Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2009, 110, 890. doi:10.1021/cr900206p |
| 7. | Driver, T. G. Org. Biomol. Chem. 2010, 8, 3831. doi:10.1039/c005219c |
| 8. | Xu, L.-M.; Li, B.-J.; Yang, Z.; Shi, Z.-J. Chem. Soc. Rev. 2010, 39, 712. doi:10.1039/B809912J |
| 9. | Asay, M.; Jones, C.; Driess, M. Chem. Rev. 2010, 111, 354. doi:10.1021/cr100216y |
| 10. | Nomura, K.; Zhang, S. Chem. Rev. 2010, 111, 2342. doi:10.1021/cr100207h |
| 11. | Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824. doi:10.1021/cr9003242 |
| 12. | Delferro, M.; Marks, T. J. Chem. Rev. 2011, 111, 2450. doi:10.1021/cr1003634 |
| 13. | Partyka, D. V. Chem. Rev. 2011, 111, 1529. doi:10.1021/cr1002276 |
| 14. | Takeuchi, D. Macromol. Chem. Phys. 2011, 212, 1545. doi:10.1002/macp.201100182 |
| 15. | Gephart, R. T., III; Warren, T. H. Organometallics 2012, 31, 7728. doi:10.1021/om300840z |
| 16. | Lang, H.; Jakob, A.; Milde, B. Organometallics 2012, 31, 7661. doi:10.1021/om300628g |
| 17. | Li, T.; Schulz, S.; Roesky, P. W. Chem. Soc. Rev. 2012, 41, 3759. doi:10.1039/c2cs15343b |
| 20. | Masuda, J. D.; Stephan, D. W. Can. J. Chem. 2005, 83, 477. doi:10.1139/v05-057 |
| 21. | Masuda, J. D.; Stephan, D. W. Dalton Trans. 2006, 2089. doi:10.1039/B513531A |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 21. | Masuda, J. D.; Stephan, D. W. Dalton Trans. 2006, 2089. doi:10.1039/B513531A |
| 22. | Cotton, F. A.; Daniels, L. M.; Matonic, J. H.; Wang, X. P.; Murillo, C. A. Polyhedron 1997, 16, 1177. doi:10.1016/s0277-5387(96)00366-x |
| 37. | Caris, R.; Peoples, B. C.; Valderrama, M.; Wu, G.; Rojas, R. J. Organomet. Chem. 2009, 694, 1795. doi:10.1016/j.jorganchem.2009.01.005 |
| 20. | Masuda, J. D.; Stephan, D. W. Can. J. Chem. 2005, 83, 477. doi:10.1139/v05-057 |
| 21. | Masuda, J. D.; Stephan, D. W. Dalton Trans. 2006, 2089. doi:10.1039/B513531A |
| 28. | Luis, S. V.; Carcia-Verdugo, E., Eds. Chemical Reactions and Processes under Flow Conditions; Royal Society of Chemistry: Cambridge, 2010. doi:10.1039/9781847559739 |
| 29. | McMullen, J. P.; Jensen, K. F. Annu. Rev. Anal. Chem. 2010, 3, 19. doi:10.1146/annurev.anchem.111808.073718 |
| 30. | Fuse, S.; Tanabe, N.; Yoshida, M.; Yoshida, H.; Doi, T.; Takahashi, T. Chem. Commun. 2010, 46, 8722. doi:10.1039/c0cc02239j |
| 31. | Suga, S.; Yamada, D.; Yoshida, J. Chem. Lett. 2010, 39, 404. doi:10.1246/cl.2010.404 |
| 32. | Baumann, M.; Baxendale, I. R.; Ley, S. V. Mol. Diversity 2011, 15, 613. doi:10.1007/s11030-010-9282-1 |
| 33. | Yoshida, J.-i.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331. doi:10.1002/cssc.201000271 |
| 34. | Fuse, S.; Tanabe, N.; Takahashi, T. Chem. Commun. 2011, 47, 12661. doi:10.1039/c1cc15662d |
| 35. | Fuse, S.; Mifune, Y.; Tanabe, N.; Takahashi, T. Org. Biomol. Chem. 2012, 10, 5205. doi:10.1039/c2ob25511a |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 36. | CCDC 934692 (3b), CCDC 934694 (3c), and 934693 (12) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 25. | Cooper, F. C.; Partridge, M. W.; Short, W. F. J. Chem. Soc. 1951, 391. doi:10.1039/jr9510000391 |
| 25. | Cooper, F. C.; Partridge, M. W.; Short, W. F. J. Chem. Soc. 1951, 391. doi:10.1039/jr9510000391 |
| 25. | Cooper, F. C.; Partridge, M. W.; Short, W. F. J. Chem. Soc. 1951, 391. doi:10.1039/jr9510000391 |
| 26. | Tanaka, K.; Motomatsu, S.; Koyama, K.; Tanaka, S.-i.; Fukase, K. Org. Lett. 2006, 9, 299. doi:10.1021/ol062777o |
| 27. | Nagaki, A.; Togai, M.; Suga, S.; Aoki, N.; Mae, K.; Yoshida, J.-i. J. Am. Chem. Soc. 2005, 127, 11666. doi:10.1021/ja0527424 |
| 23. | Fuse, S.; Masui, H.; Tannna, A.; Shimizu, F.; Takahashi, T. ACS Comb. Sci. 2012, 14, 17. doi:10.1021/co200081j |
| 24. | Masui, H.; Fuse, S.; Takahashi, T. Org. Lett. 2012, 14, 4090. doi:10.1021/ol3017337 |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 19. | Peoples, B. C.; De la Vega, G.; Valdebenito, C.; Quijada, R.; Ibañez, A.; Valderrama, M.; Rojas, R. J. Organomet. Chem. 2012, 700, 147. doi:10.1016/j.jorganchem.2011.11.035 |
| 18. | Valdebenito, C.; Garland, M. T.; Quijada, R.; Rojas, R. J. Organomet. Chem. 2009, 694, 717. doi:10.1016/j.jorganchem.2008.11.066 |
| 21. | Masuda, J. D.; Stephan, D. W. Dalton Trans. 2006, 2089. doi:10.1039/B513531A |
| 39. | MMAO is a modified methylaluminoxane activator containing 25% isobutyl aluminoxane prepared by the controlled hydrolysis of Me3Al and (iBu)3Al. |
© 2013 Fuse et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)