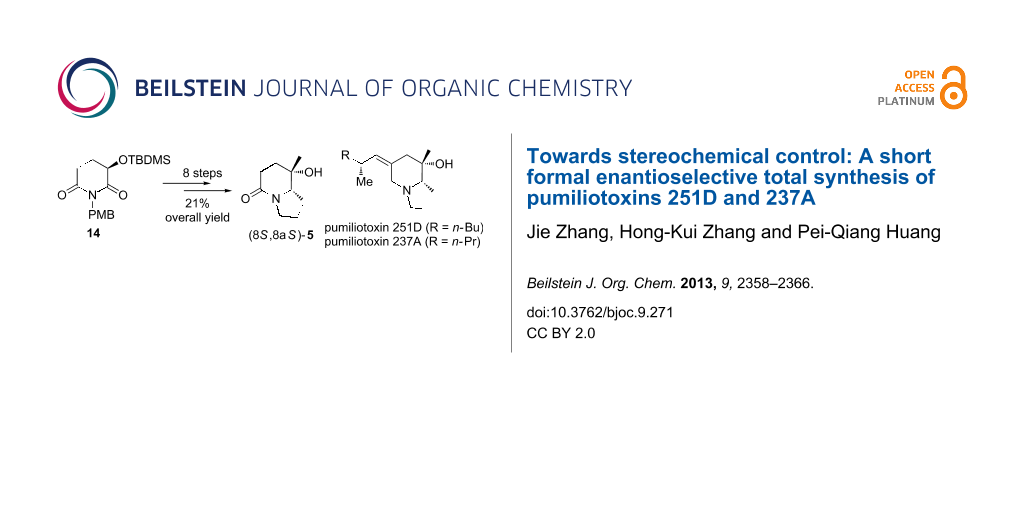
Abstract
A concise enantioselective synthesis of the advanced intermediate 5 for the synthesis of pumiliotoxins (Gallagher’s intermediate) is described. The synthesis started from the regio- and trans-diastereoselective (dr = 98:2) reductive 3-butenylation of (R)-3-(tert-butyldimethylsilyloxy)glutarimide 14. After O-desilylation and Dess–Martin oxidation, the resulting keto-lactam 10 was subjected to a highly trans-stereoselective addition of the methylmagnesium iodide to give carbinol 11 as sole diastereomer. An efficient ring closure procedure consisting of ozonolysis and reductive dehydroxylation provided the indolizidine derivative 5, which completed the formal enantioselective total synthesis of pumiliotoxins 251D and 237A.

Graphical Abstract
Introduction
Pumiliotoxins (PTXs, 1, Figure 1) such as pumiliotoxin 251D (2) are a subclass of indolizidine alkaloids isolated from the skin secretion of neotropical frogs. A total of 19 members have been isolated and partially characterized [1]. Pumiliotoxins are structurally characterized by a (Z)-6-alkylidene-8-hydroxy-8-methylindolizidine ring system, which distinguishes from one to another by the 6-alkylidene side chain [1]. Interestingly, it is known that poison frogs don’t produce the alkaloids themselves, instead, they accumulate alkaloids from dietary alkaloid-containing arthropods serving as a chemical defense against predators. It is not surprising that the alkaloids found in poison frogs can also be detected from ants, and most of them show remarkable bioactivities [2,3]. For example, formicine ants have been shown to be an arthropod source of the pumiliotoxin alkaloids of dendrobatid poison frogs [4,5]. Forthermore, from the extracts of an unidentified Scheloribates sp. of mites, pumiliotoxin 237A (3) was detected as a minor component [6].
![[1860-5397-9-271-1]](/bjoc/content/figures/1860-5397-9-271-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of some pumiliotoxins and an advanced intermediate.
Figure 1: Structures of some pumiliotoxins and an advanced intermediate.
That the wide range of biological activities possessed by these molecules have attracted much attention of synthetic organic chemists, and numerous approaches have been reported [7-9]. Pumiliotoxin 251D (PTX 251D) (2) is the first structurally defined member of pumiliotoxins, a class of dendrobatid alkaloids isolated from Ecuadorean poisonous frog, Dendrobares tricolor in 1980 [10]. Since the pioneering work of Overman [11], a number of approaches have been developed for the synthesis of PTX 251D [12-26]. Among them, that of Gallagher [12] has attracted much attention. They demonstrated that (8S,8aS)-8-hydroxy-8-methyl-5-indolizidinone (5, Figure 1) can serve as an advanced intermediate for the synthesis of pumiliotoxin 251D [12]. Later on, Nubbemeyer and co-workers used the Horner olefination to convert 5 and its diastereomer into (+)-PTX 251D (2) and the 8-epimer of PTX 209F (4), respectively [17]. Recently, (8S,8aS)-5 has been used for the synthesis of PTX 237A (3) [6]. Thus, it is logic to envision that an efficient synthesis of this key intermediate would allow access to other member of pumiliotoxins and their analogues. To date more than ten synthetic approaches to (8S,8aS)-5 have been published [12-15,18-22,27-31], among them L-proline and its derivative were used as popular precursors from the pool of chiral compounds [13-15,18-22]. Herein, we report a concise diastereoselective synthesis of (8S,8aS)-5 starting from (R)-3-(tert-butyldimethylsilyloxy)glutarimide 14, a versatile building block developed from our laboratory [32].
Results and Discussion
In recent years, we have been engaged in the development of efficient methodologies for the synthesis of nitrogen-containing bioactive heterocycles [33-37], and recently reported an approach for the synthesis of (8R,8aS)-8-hydroxy-5-indolizidinone (6) [38]. As a continuation of that study, the synthesis of (8S,8aS)-5 starting from compound 6 was envisioned. For this purpose, alcohol 6 was subjected to Swern oxidation (DMSO, (COCl)2, iPr2NEt, −78 to 0 °C, 3 h), and the resulting keto-lactam 7 treated with MeMgI in diethyl ether (−78 to 0 °C, 1 h), which gave alcohols 5/5a as an inseparable mixture of diastereomers in a ratio of 1:2.2 (combined yield: 47% over 2 steps) (Scheme 1). A comparison of spectral data (1H and 13C NMR) of our products 5/5a with those reported [12,22,28] showed that the desired (8S,8aS)-5 was the minor diastereomer. Similar results have been reported by Nubbemeyer [27] and Li [8]. In view of the fruitless efforts of Nubbemeyer [27] and Li [8] in inversing the diastereoselectivity of the methylation reaction of keto-lactam 7, an alternative approach was envisaged.
![[1860-5397-9-271-i1]](/bjoc/content/inline/1860-5397-9-271-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 5 from 6 via oxidation–addition sequence.
Scheme 1: Synthesis of 5 from 6 via oxidation–addition sequence.
To develop a diastereoselective approach to (8S,8aS)-5, it would be helpful to analyze the plausible stereochemical course of the methylation of keto-lactam 7. The observed unusual stereoselective cis-methylation implicates a preferential axial attack of the methylmagnesium iodide to the bicyclic keto-lactam 7 (Scheme 2). Although the preferential axial attack of “small” nucleophiles to cyclohexanone is well known [39-41], the “small” nucleophiles are limited to some metallo-hydrides, metallo-acetylenide [42] and (cyanomethyl)lithium [43]. In the case of bicyclic keto-lactam 7, presumably, the equatorial attack of methylmagnesium iodide to give 5, suffers from unfavorable eclipsing interactions between the incoming methylmagnesium iodide and the vicinal methylene group. As a consequence, the axial attack to give 5a is more favorable.
![[1860-5397-9-271-i2]](/bjoc/content/inline/1860-5397-9-271-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible stereochemical course of the preferential axial addition of methylmagnesium iodide to bicyclic keto-lactam 7.
Scheme 2: Plausible stereochemical course of the preferential axial addition of methylmagnesium iodide to bic...
It was envisioned that if a monocyclic keto-lactam was used, the situation would be changed and an equatorial attack of the nucleophile giving the cis-product would be preferable. Indeed, Holmes and co-workers have reported that methylmagnesium bromide addition to N-Cbz-protected piperidin-3-one 8 produced exclusively the trans-methylation product 9 [44] (Scheme 3). In that case, the N-benzyloxycarbonyl group imposed A1,3-strain on piperidine derivatives founded the basis for the observed stereocontrol.
![[1860-5397-9-271-i3]](/bjoc/content/inline/1860-5397-9-271-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Holmes’ exclusive trans-diastereoselective methylation of N-Cbz-protected piperidin-3-one 8.
Scheme 3: Holmes’ exclusive trans-diastereoselective methylation of N-Cbz-protected piperidin-3-one 8.
Thus, we chose keto-lactam 10 as our substrate. Although compound 10 is not a urethane, and a A1,3-strain not longer exists, a simple chair conformational-controlled preferential equatorial attack could be expected (Scheme 4). Substrate 10 may adopt two plausible chair conformations 10a and 10b, in which conformation 10b with a pseudoequatorially positioned N-α-substitutent is more stable. Thus, the pseudoequatorial attack of MeMgI would be preferential, which gives the product 11 as the major product.
![[1860-5397-9-271-i4]](/bjoc/content/inline/1860-5397-9-271-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Our plan for the trans-diastereoselective methylation of keto-lactam 10.
Scheme 4: Our plan for the trans-diastereoselective methylation of keto-lactam 10.
On the basis of the abovementioned analysis, a retrosynthetic analysis of (8S,8aS)-5 is displayed in Scheme 5, which features the formation of the fused pyrrolidine ring from the but-3-ene-1-yl group, the expected trans-diastereoselective methylation as the key step, and protected (R)-3-hydroxyglutarimide-based regio- and trans-diastereoselective reductive alkylation, a synthetic methodology developed from our laboratory [32,38,45,46], as the starting point.
![[1860-5397-9-271-i5]](/bjoc/content/inline/1860-5397-9-271-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Retrosynthetic analysis of (8S,8aS)-8-hydroxy-8-methylindolizidin-5-one (5).
Scheme 5: Retrosynthetic analysis of (8S,8aS)-8-hydroxy-8-methylindolizidin-5-one (5).
Our synthesis started from the addition of a freshly prepared 3-butenylmagnesium bromide to imide 13 [47] in dichloromethane at −78 °C for 3 h, which gave regioselective carbinol 15 as a mixture of two diastereomers. Without separation, the mixture was treated with Et3SiH/BF3·Et2O (−78 °C to rt, 2 h) [32] to yield the reductive dehydroxylation products 16 and 17 in a ratio of 98:2 (determined by 1H NMR) [32] (combined yield: 88% over 2 steps), from which only the major product 16 was isolated (Scheme 6). For the chemoselective debenzylation, lithium naphthalenide (LN) was used [48]. To our disappointment, attempted cleavage of the benzyl group by LN (THF, −40 °C to rt, 2 h) gave the desired 18 in only 20% yield along with 60% of the recovered starting material 16.
![[1860-5397-9-271-i6]](/bjoc/content/inline/1860-5397-9-271-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Due to the low yield in the debenzylation process, we decided to change the O-protecting group from benzyl to TBDMS, namely, use of TBDMS ether 14 as the starting material. Thus, (R)-3-(tert-butyldimethylsilyloxy)glutarimide 14 [49] was prepared from the known (R)-3-hydroxyglutarimide 19 (prepared from (R)-glutamic acid in 69% overall yield over 4 steps [47]) in a yield of 95%. Successive treatment of imide 14 with 3-butenylmagnesium bromide (CH2Cl2, −20 °C, 3 h) and the resulting hemi-aminal with Et3SiH/BF3·Et2O (CH2Cl2, −78 °C, 2 h, then −20 °C, 2 h) gave a diastereomeric mixture of lactams. Without separation, the mixture was treated with TBAF in dichloromethane to produce alcohols 18 and 20 (combined yield: 62% over 3 steps) in a ratio of 95:5 (determined by 1H NMR), along with the keto-amide 21 in 13% yield (Scheme 7). Separation of the mixture by column chromatography gave pure diastereomer 18 in 59% overall yield from 14. The trans-stereochemistry was tentatively assigned to compound 18 on the basis of our previous results, which was confirmed by the conversion of 18 into the known compound 5 (vide infra).
![[1860-5397-9-271-i7]](/bjoc/content/inline/1860-5397-9-271-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
With lactam 18 in hand, its conversion to novel tertiary alcohol 22 was investigated. Oxidation of alcohol 18 with an excess of Dess–Martin periodinane (DMP) in dichloromethane at room temperature for 2 h proceeded smoothly to give ketone 10 in 93% yield. It is noteworthy that keto-lactam 10 is configurational labile, and should be used immediately in the next reaction. Treatment of keto-lactam 10 with MeMgI (3 equiv) in dichloromethane (0 °C to rt, 12 h) gave a 76% yield of the desired methylation product 11 as the sole observable diastereomer (Scheme 8), whose stereochemistry was determined after converting into the known (8S,8aS)-5. This result verifies the previous assumption outlined in Scheme 4.
![[1860-5397-9-271-i8]](/bjoc/content/inline/1860-5397-9-271-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of tertiary alcohol 22.
Scheme 8: Synthesis of tertiary alcohol 22.
To remove the PMB protecting group, compound 11 was treated with ammonium cerium nitrate (CAN) in CH3CN/H2O (v/v 3:1) [50] at room temperature for 4 h to afford the deprotected lactam 22 in 56% yield.
Our next task was the oxidative cyclization of the olefin 22 to form the indolizidinone ring. Thus, ozonolysis of olefin 22 in dichloromethane [51], followed by quenching with Me2S furnished the hemiaminal tautomer via intermediacy of lactam-aldehyde. Without isolation, the crude was subjected to the reductive dehydroxylation with Et3SiH/BF3·Et2O (CH2Cl2, −78 °C) to give the desired indolizidinone 5 in an overall yield of 91% from 22 (Scheme 9).
![[1860-5397-9-271-i9]](/bjoc/content/inline/1860-5397-9-271-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of (8S,8aS)-5 and its silyl ether 23.
Scheme 9: Synthesis of (8S,8aS)-5 and its silyl ether 23.
The spectral data of the our synthetic (8S,8aS)-5 are in agreement with those reported [12,22,27-29]. Due to the differences between the reported optical rotation values {[α]D20 −42.7 (c 1.0, CHCl3); lit. [12] [α]D21 −47.0 (c 0.97, CHCl3); lit. [22] [α]D22 −55 (c 0.79, CHCl3); lit. [28] [α]D25 −41.3 (c 0.48, CHCl3); lit. [29] [α]D25 −32.1 (c 1.0, CHCl3)}, compound 5 was converted into the known silyl ether 23 (TMSCl, imidazole, DMAP, CH2Cl2, 0 °C to rt, 24 h, 91% yield), which is identical in all aspects with that reported by Nubbemeyer {[α]D20 −35.3 (c 0.89, CHCl3); lit. [27] [α]D20 −35.1 (c 1.08, CHCl3)}. Silyl ether 23 may serve as an advanced intermediate in the synthesis of pumiliotoxin 209F [17].
Conclusion
In summary, an eight-step synthesis of the advanced intermediate (8S,8aS)-5 for the syntheis of pumiliotoxins has been achieved in 21% overall yield starting from (R)-3-silyloxyglutarimide derivative 14. The method is based on the versatile building block 14 and relied on a highly diastereoselective trans-addition of methylmagnesium iodide to keto-lactam 10. Since compound (8S,8aS)-5 has been converted into pumiliotoxins 251D (2) and 237A (3), by Gallagher, Nubbemeyer, and Mori, respectively, its synthesis constitutes a formal enantioselective total synthesis of pumiliotoxins 251D and 237A. Compound (8S,8aS)-5 would be of value for the synthesis of other pumiliotoxins as well. The highly diastereoselective trans-addition of a methylmagnesium iodide to keto-lactam 10 provides a new example of achieving the desired diastereoselection simply by the chair-conformation control [52,53].
Experimental
Optical rotations were recorded on a Perkin-Elmer 341 automatic polarimeter. 1H NMR and 13C NMR spectra were recorded on bruker 400 spectrometer. 1H NMR spectra were measured in CDCl3, and chemical shifts are expressed in parts per million (ppm) relative to internal Me4Si. IR spectra were recorded on a Nicolet Avatar 330 RT-IR spectrophotometer. Mass spectra were recorded by Bruker Dalton Esquire 3000 plus and Finnigan Mat-LCQ (ESI direct injection). HRFABMS spectra were recorded on a Bruker APEX-FTMS apparatus. Elemental analyses were performed using a Vario RL analyzer. Melting points were determined on a Yanaco MP-500 melting point apparatus and are uncorrected.
Tetrahydrofuran (THF) was distilled prior to use from sodium benzophenone ketyl. Dichloromethane was distilled from phosphorus pentoxide. Silica gel (zhifu, 300–400 mesh) from Yantai silica gel factory (China) was used for column chromatography, eluting (unless otherwise stated) with ethyl acetate/petroleum ether (PE) (60–90 °C) mixture.
(R)-3-(tert-Butyldimethylsilyloxy)-1-(4-methoxybenzyl)piperidine-2,6-dione (14): To a solution of (R)-3-hydroxy-1-(4-methoxybenzyl)piperidine-2,6-dione 19 [47] (125 mg, 0.5 mmol), DMAP (10 mg) and imidazole (67 mg, 1 mmol) in CH2Cl2 (15 mL) was added TBDMSCl (52 μL, 0.6 mmol). The mixture was stirred at room temperature for 24 h before quenching with H2O (5 mL). The organic layer was separated and the aqueous phase extracted with CH2Cl2 (10 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane 1:2) to give silyl ether 14 (517 mg, yield: 95%) as a colorless oil. [α]D20 +19.2 (c 1.1, CHCl3); IR (film) νmax: 3062, 2936, 1728, 1658, 1494, 1451, 1367 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.11 (s, 3H), 0.12 (s, 3H), 0.87 (s, 9H), 1.92–2.07 (m, 2H), 2.59 (ddd, J = 17.7, 7.5, 5.4 Hz, 1H), 2.91 (ddd, J = 17.7, 8.1, 5.4 Hz, 1H), 3.76 (s, 3H), 4.30 (dd, J = 7.4, 4.0 Hz, 1H), 4.83 (d, J = 14.0 Hz, 1H), 4.86 (d, J = 14.0 Hz, 1H), 6.79 (d, J = 8.7 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ −5.4, −4.7, 18.2, 25.6 (3C), 26.4, 29.1, 42.4, 55.2, 69.3, 113.7 (2C), 129.4, 130.3 (2C), 158.9, 171.8, 172.2; HRMS ESI (m/z): [M + Na]+ calcd for C19H29NO4SiNa, 386.1758; found, 386.1757.
(5R,6S)-6-(But-3-enyl)-5-hydroxy-1-(4-methoxybenzyl)piperidin-2-one (18): To a solution of compound 14 (2.8 g, 7.71 mmol) in anhydrous CH2Cl2 (100 mL) at −20 °C was added dropwise a freshly prepared 3-butenylmagnesium bromide (1 M in THF, 15 mL, 15 mmol). After being stirred at −20 °C for 3 h, the reaction was quenched with a saturated aqueous solution of NH4Cl (15 mL). The organic layer was separated and the aqueous phase was extracted with CH2Cl2 (25 mL × 5). The combined organic layers were washed with brine (10 mL × 5), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Without further separation, the crude product (2.4 g, 5.7 mmol) was treated with Et3SiH (9 mL, 57 mmol) and BF3·Et2O (2.12 mL, 17.2 mmol) in CH2Cl2 (100 mL) at −78 °C. The mixture was stirred at −78 °C for 2 h, and then allowed to warm to −20 °C. After being stirred for another 2 h, saturated aqueous solution of NaHCO3 (25 mL) was added and aqueous layer was extracted with CH2Cl2 (15 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a crude product, which was used in the next step without further purification. To a solution of the crude product in THF (50 mL) was added a 1 M solution of TBAF in THF (17.1 mL, 17.1 mmol) at 0 °C. The mixture was allowed to warm to room temperature. After being stirred for 4 h at room temperature, the reaction was quenched with water, and extracted with CH2Cl2 (15 mL × 5). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane 2:1) to give diastereomers 18 (969 mg, yield: 59%) and 20 (51 mg, yield: 3%). Compound 18: white solid, mp 102–105 °C (EtOAc/hexane); [α]D20 −65.0 (c 1.0, CHCl3); IR (film) νmax: 3375, 2934, 1612, 1512, 1193, 1056, 784, 560 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.37–1.49 (m, 1H), 1.67–1.64 (m, 2H), 1.91–2.00 (m, 2H), 2.28 (ddd, J = 18.1, 7.2, 2.8 Hz, 1H), 2.58 (ddd, J = 18.1, 10.5, 7.6 Hz, 2H), 3.20 (dt, J = 9.4, 2.6 Hz, 1H), 3.72 (s, 3H), 3.81 (d, J = 14.9 Hz, 1H), 3.93 (dt, J = 4.6, 2.6 Hz, 1H), 4.91–5.01 (m, 2H), 5.14 (d, J = 14.9 Hz, 1H), 5.69 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H), 6.78 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 23.9, 26.7, 30.0, 31.3, 47.3, 55.1, 61.8, 65.0, 113.8 (2C), 115.5, 129.0, 129.1 (2C), 136.9, 158.7, 169.8; HRMS ESI (m/z): [M + Na]+ calcd for C17H23NO3Na, 312.1570; found, 312.1570.
(S)-6-(But-3-en-1-yl)-1-(4-methoxybenzyl)piperidine-2,5-dione (10): To a stirred solution of compound 18 (100 mg, 0.34 mmol) in CH2Cl2 (5 mL) was added Dess–Martin periodinane (220 mg, 0.52 mmol) at room temperature. After being stirred for 2 h, the reaction was quenched with a 10% aqueous solution of Na2S2O3. The aqueous phase was extracted with EtOAc (15 mL × 3). The combined organic layers were washed successively with a saturated aqueous solution of NaHCO3 (5 mL × 3) and brine (5 mL × 2). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane 1:2) to give compound 10 (98 mg, yield: 93%) as a colorless oil. [α]D20 +37.5 (c 1.0, CHCl3); IR (film) νmax: 2955, 2926, 2834, 1725, 1625, 1512, 1246, 1173, 1032 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.79–2.11 (m, 4H), 2.57–2.69 (m, 2H), 2.73–2.80 (m, 2H), 3.68 (dd, J = 7.5, 4.6 Hz, 1H), 3.79 (s, 3H), 3.85 (d, J = 14.7 Hz, 1H), 4.97–5.08 (m, 2H), 5.28 (d, J = 14.7 Hz, 1H), 5.70 (ddt, J = 16.7, 10.2, 6.4 Hz, 1H), 6.84 (d, J = 8.7 Hz, 2H), 7.15 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 29.0, 29.2, 30.6, 35.3, 47.2, 55.2, 63.9, 114.2 (2C), 116.3, 128.3, 129.5 (2C), 136.4, 159.3, 169.6, 206.2; HRMS ESI (m/z): [M + Na]+ calcd for C17H21NO3Na, 310.1414; found, 310.1419.
(5S,6S)-6-(But-3-enyl)-5-hydroxy-1-(4-methoxybenzyl)-5-methylpiperidin-2-one (11): To a solution of compound 10 (92 mg, 0.32 mmol) in anhydrous CH2Cl2 (100 mL) was added dropwise a freshly prepared 1 M diethyl ether solution of CH3MgI (1.0 mL, 1.0 mmol) at 0 °C. After being stirred at room temperature overnight, the reaction was quenched with a saturated aqueous solution of NH4Cl (5 mL). The organic layer was separated and the aqueous phase was extracted with CH2Cl2 (15 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane 1:1) to give compound 11 (75 mg, yield: 76%) as a colorless oil. [α]D20 −73.5 (c 1.0, CHCl3); IR (film) νmax: 3378, 2928, 2925, 2874, 1612, 1512, 1247, 1150, 1034, 914, 847 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.99 (s, 3H), 1.57–1.65 (m, 2H), 1.73 (d, J = 16.5 Hz, 1H), 2.04–2.12 (m, 2H), 2.20–2.27 (m, 2H), 2.41 (ddd, J = 18.6, 9.5, 8.8 Hz, 1H), 2.55 (ddd, J = 18.6, 8.8, 2.2 Hz, 1H), 3.00 (td, J = 5.4, 1.6 Hz, 1H), 3.59 (d, J = 14.3 Hz, 1H), 3.78 (s, 3H), 5.00–5.10 (m, 2H), 5.45 (d, J = 14.3 Hz, 1H), 5.81 (ddt, J = 16.8, 10.2, 6.7 Hz, 1H), 6.83 (d, J = 8.6 Hz, 2H), 7.20 (d, J = 8.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 26.9, 29.0, 30.5, 30.8, 32.5, 48.6, 55.2, 63.1, 70.1, 113.8 (2C), 115.5, 129.0, 130.4 (2C), 138.0, 159.1, 168.7; HRMS ESI (m/z): [M + Na]+ calcd for C18H25NO3Na, 326.1727; found, 326.1728.
(5S,6S)-6-(But-3-enyl)-5-hydroxy-5-methylpiperidin-2-one (22): To a solution of compound 11 (463 mg, 1.52 mmol) in a mixture of CH3CN (32 mL) and H2O (11 mL) was added ammonium cerium nitrate (2.5 g, 4.56 mmol) in one portion. The mixture was stirred for 4 h at room temperature. To the resulting mixture was added H2O (5 mL), and the mixture was extracted with EtOAc (30 mL × 5). The combined organic layers were washed successively with a saturated solution aqueous of NaHCO3 and brine. The organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 1:40) to give compound 22 (153 mg, yield: 56%) as a pale yellow oil. [α]D20 −43.0 (c 1.18, CHCl3); IR (film) νmax: 3366, 2932, 1612, 1475, 1406, 1312, 919 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.31 (s, 3H), 1.52–1.61 (m, 1H), 1.76–1.93 (m, 3H), 2.10 (m, 1H), 2.22–2.35 (m, 2H), 2.57 (ddd, J = 18.2, 11.1, 7.0 Hz, 1H), 2.80 (s, 1H, OH, D2O exchangeable), 3.17 (dd, J = 10.2, 1.8 Hz, 1H), 5.03–5.12 (m, 2H), 5.79 (ddt, J = 17.0, 10.2, 6.6 Hz, 1H), 6.03 (s, 1H, NH); 13C NMR (100 MHz, CDCl3) δ 26.3, 27.5, 28.7, 30.0, 34.1, 60.1, 67.9, 115.9, 137.1, 172.4; HRMS ESI (m/z): [M + Na]+ calcd for C10H17NO2Na, 206.1151; found, 206.1160.
(8S,8aS)-8-Hydroxy-8-methyloctahydroindolizidin-5-one (5): To a stirred solution of compound 22 (90 mg, 0.49 mmol) in a mixture of CH2Cl2 (8 mL) and MeOH (2 mL) was bubbled O3 at −78 °C for 10 min. The reaction was quenched with Me2S (0.2 mL). The mixture was allowed to warm to room temperature. The organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (15 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Without further purification, to a solution of the crude mixture in CH2Cl2 (5 mL) was added Et3SiH (0.77 mL, 4.9 mmol) and BF3·Et2O (0.18 mL, 1.47 mmol) at −78 °C. The mixture was allowed to warm slowly to the room temperature. A saturated aqueous solution of NaHCO3 (2 mL) was added and aqueous phase was extracted with CH2Cl2 (15 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (MeOH/CH2Cl2 1:20) to give compound 5 (75 mg, yield: 91%) as a colorless waxy solid. [α]D20 −42.7 (c 1.0, CHCl3) {lit. [12] [α]D20 −47.0 (c 0.8, CHCl3) lit. [28] [α]D25 −41.3 (c 0.48, CHCl3); lit. [29] [α]D25 −32.1 (c 1.0, CHCl3)}; IR (film) νmax: 3364, 2926, 2877, 1612, 1469, 1265, 740, 703 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.29 (s, 3H), 1.74–1.97 (m, 6H), 2.20 (br s, 1H, OH, D2O exchangeable), 2.38 (dd, J = 18.4, 7.4 Hz, 1H), 2.53 (ddd, J = 18.4, 11.7, 7.4 Hz, 1H), 3.35 (dd, J = 10.3, 5.3 Hz, 1H), 3.48–3.53 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 22.0, 26.3, 26.4, 28.0, 35.1, 45.7, 66.1, 67.7, 169.0; HRMS ESI (m/z): [M + Na]+ calcd for C9H15NO2Na, 192.0995; found, 192.0999.
(8S,8aS)-8-Methyl-8-trimethylsilyloxyoctahydroindolizidin-5-one (23): TMSCl (25 μL, 0.28 mmol) was added a solution of compound 5 (40 mg, 0.24 mmol), DMAP (5 mg) and imidazole (32 mg, 0.48 mmol) in CH2Cl2 (8 mL) at 0 °C. The mixture was stirred at room temperature for 24 h, and then diluted with H2O (2 mL). The organic layer was separated and the aqueous phase was extracted with CH2Cl2 (5 mL × 5). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane 1:2) to give compound 23 (53 mg, yield: 91%) as a pale yellow oil. [α]D20 −35.3 (c 0.89, CHCl3) {lit. [27] [α]D20 −35.1 (c 1.08, CHCl3)}; IR (film) νmax: 2955, 2880, 1621, 1470, 1413, 1378, 1316, 1273, 1265, 1253, 1224, 1134, 1068, 1021 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.05 (s, 9H), 1.26 (s, 3H), 1.60–1.90 (m, 6H), 2.25–2.33 (m, 1H), 2.33–2.45 (m, 1H), 3.15–3.20 (dd, J = 10.0, 5.5 Hz, 1H), 3.38–3.48 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 2.1, 21.9, 26.2, 26.3 , 28.2, 35.2, 45.7, 67.3, 70.4, 168.9; HRMS ESI (m/z): [M + Na]+ calcd for C12H23NO2SiNa , 264.1390; found, 264.1392.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR of key compounds. | ||
| Format: PDF | Size: 552.1 KB | Download |
Acknowledgments
The authors are grateful to National Basic Research Program (973 Program) of China (Grant No. 2010CB833200), the NSF of China (21072160; 21332007), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of Ministry of Education, China for financial support. We thank Mr. Xin Li for the initial work displayed in Scheme 1.
References
-
Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556–1575. doi:10.1021/np0580560
Return to citation in text: [1] [2] -
Vandendriessche, T.; Abdel-Mottaleb, Y.; Maertens, C.; Cuypers, E.; Sudau, A.; Nubbemeyer, U.; Mebs, D.; Tytgat, J. Toxicon 2008, 51, 334–344. doi:10.1016/j.toxicon.2007.10.011
Return to citation in text: [1] -
Daly, J. W.; Gusovsky, F.; McNeal, E. T.; Secunda, S.; Bell, M.; Creveling, C. R.; Nichizawa, Y.; Overman, L. E.; Sharp, M. J.; Rossignol, D. P. Biochem. Pharmacol. 1990, 40, 315–326. doi:10.1016/0006-2952(90)90694-G
Return to citation in text: [1] -
Smith, S. Q.; Jones, T. H. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7841–7842. doi:10.1073/pnas.0402599101
Return to citation in text: [1] -
Saporito, R. A.; Garraffo, H. M.; Donnelly, M. A.; Edwards, A. L.; Longino, J. T.; Daly, J. W. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8045–8050. doi:10.1073/pnas.0402365101
Return to citation in text: [1] -
Takada, W.; Sakata, T.; Shimano, S.; Enami, Y.; Mori, N.; Nishida, R.; Kuwahara, Y. J. Chem. Ecol. 2005, 31, 2403–2415. doi:10.1007/s10886-005-7109-9
Return to citation in text: [1] [2] -
Kibayashi, C. Chem. Pharm. Bull. 2005, 53, 1375–1386. doi:10.1248/cpb.53.1375
Return to citation in text: [1] -
Tang, B. C.; Li, W. D. Chin. J. Org. Chem. 2004, 24, 1151–1158.
Return to citation in text: [1] [2] [3] -
Franklin, A. S.; Overman, L. E. Chem. Rev. 1996, 96, 505–522. doi:10.1021/CR950021P
Return to citation in text: [1] -
Daly, J. W.; Tokuyama, T.; Fujiwara, T.; Higher, R. J.; Karlet, I. L. J. Am. Chem. Soc. 1980, 102, 830–836. doi:10.1021/ja00522a064
Return to citation in text: [1] -
Overman, L. E.; Bell, K. L. J. Am. Chem. Soc. 1981, 103, 1851–1853. doi:10.1021/ja00397a052
Return to citation in text: [1] -
Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Pinho, V. D.; Procter, D. J.; Burtoloso, A. C. B. Org. Lett. 2013, 15, 2434–2437. doi:10.1021/ol400903n
Return to citation in text: [1] [2] [3] -
Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w
Return to citation in text: [1] [2] [3] -
Sultane, P. R.; Mohite, A. R.; Bhat, R. G. Tetrahedron Lett. 2012, 53, 5856–5858. doi:10.1016/j.tetlet.2012.08.061
Return to citation in text: [1] [2] [3] -
Woodin, K. S.; Jamison, T. F. J. Org. Chem. 2007, 72, 7451–7454. doi:10.1021/jo071132e
Return to citation in text: [1] -
Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3315–3325. doi:10.1002/1099-0690(200210)2002:19<3315::AID-EJOC3315>3.0.CO;2-2
Return to citation in text: [1] [2] [3] -
Ni, Y.; Zhao, G.; Ding, Y. J. Chem. Soc., Perkin Trans. 1 2000, 3264–3266. doi:10.1039/b004450o
Return to citation in text: [1] [2] [3] -
Barrett, A. G. M.; Damiani, F. J. Org. Chem. 1999, 64, 1410–1411. doi:10.1021/jo9820972
Return to citation in text: [1] [2] [3] -
Cossy, J.; Cases, M.; Gomez Pardo, D. Bull. Soc. Chim. Fr. 1997, 134, 141–144.
Return to citation in text: [1] [2] [3] -
Cossy, J.; Cases, M.; Gomez Pardo, D. Synlett 1996, 909–910. doi:10.1055/s-1996-5590
Return to citation in text: [1] [2] [3] -
Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Bargar, T. M.; Lett, R. M.; Johnson, P. L.; Hunter, J. E.; Chang, C. P.; Pernich, D. J.; Sabol, M. R.; Dick, M. R. J. Agric. Food Chem. 1995, 43, 1044–1051. doi:10.1021/jf00052a037
Return to citation in text: [1] -
Honda, T.; Hoshi, M.; Kanai, K.; Tsubuki, M. J. Chem. Soc., Perkin Trans. 1 1994, 2091–2101. doi:10.1039/p19940002091
Return to citation in text: [1] -
Honda, T.; Hoshi, M.; Tsubuki, M. Heterocycles 1992, 34, 1515–1518. doi:10.3987/COM-92-6098
Return to citation in text: [1] -
Overman, L. E.; Bell, K. L.; Ito, F. J. Am. Chem. Soc. 1984, 106, 4192–4201. doi:10.1021/ja00327a022
Return to citation in text: [1] -
Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015
Return to citation in text: [1] [2] [3] [4] [5] -
O’Mahony, G.; Nieuwenhuyzen, M.; Armstrong, P.; Stevenson, P. J. J. Org. Chem. 2004, 69, 3968–3971. doi:10.1021/jo0497205
Return to citation in text: [1] [2] [3] [4] -
Chen, B.-F.; Tasi, M.-R.; Yang, C.-Y.; Chang, J.-K.; Chang, N.-C. Tetrahedron 2004, 60, 10223–10231. doi:10.1016/j.tet.2004.09.001
Return to citation in text: [1] -
Wang, B.; Fang, F.; Lin, G.-Q. Tetrahedron Lett. 2003, 44, 7981–7984. doi:10.1016/j.tetlet.2003.08.113
Return to citation in text: [1] -
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
See for an account on the methodology of the regio-and trans-diastereoselective reductive addition of glutarimide.
Return to citation in text: [1] [2] [3] [4] -
Liu, X.-K.; Ye, J.-L.; Ruan, Y.-P.; Li, Y.-X.; Huang, P.-Q. J. Org. Chem. 2013, 78, 35–41. doi:10.1021/jo3014484
Return to citation in text: [1] -
Huo, H.-H.; Xia, X.-E.; Zhang, H.-K.; Huang, P.-Q. J. Org. Chem. 2013, 78, 455–465. doi:10.1021/jo302362b
Return to citation in text: [1] -
Liu, X.-K.; Zheng, X.; Ruan, Y.-P.; Ma, J.; Huang, P.-Q. Org. Biomol. Chem. 2012, 10, 1275–1284. doi:10.1039/c1ob06697h
Return to citation in text: [1] -
Zhuang, J.-J.; Ye, J.-L.; Zhang, H.-K.; Huang, P.-Q. Tetrahedron 2012, 68, 1750–1755. doi:10.1016/j.tet.2011.12.063
Return to citation in text: [1] -
Liu, X.-K.; Qiu, S.; Xiang, Y.-G.; Ruan, Y.-P.; Zheng, X.; Huang, P.-Q. J. Org. Chem. 2011, 76, 4952–4963. doi:10.1021/jo200600n
Return to citation in text: [1] -
Zhang, H.-K.; Li, X.; Huang, H.; Huang, P.-Q. Sci. China: Chem. 2011, 54, 737–744. doi:10.1007/s11426-011-4256-4
Return to citation in text: [1] [2] -
Wu, Y. D.; Houk, K. N.; Florez, J.; Trost, B. M. J. Org. Chem. 1991, 56, 3656–3664. doi:10.1021/jo00011a039
Return to citation in text: [1] -
Ashby, E. C.; Laemmle, J. T. Chem. Rev. 1975, 75, 521–546. doi:10.1021/cr60296a005
Return to citation in text: [1] -
Trost, B. M.; Florez, J.; Jebaratnam, D. J. J. Am. Chem. Soc. 1987, 109, 613–615. doi:10.1021/ja00236a067
Return to citation in text: [1] -
Huang, P. Q.; Zhou, W. S. Tetrahedron: Asymmetry 1991, 2, 875–878. doi:10.1016/S0957-4166(00)82199-0
Return to citation in text: [1] -
Dimitrov, V.; Panev, S. Tetrahedron: Asymmetry 2000, 11, 1513–1516. doi:10.1016/S0957-4166(00)00099-9
Return to citation in text: [1] -
Tan, C.-H.; Holmes, A. B. Chem.–Eur. J. 2001, 7, 1845–1854. doi:10.1002/1521-3765(20010504)7:9<1845::AID-CHEM1845>3.0.CO;2-2
Return to citation in text: [1] -
Liu, L.-X.; Xiao, K.-J.; Huang, P.-Q. Tetrahedron 2009, 65, 3834–3841. doi:10.1016/j.tet.2009.03.021
Return to citation in text: [1] -
Liu, L.-X.; Ruan, Y.-P.; Guo, Z.-Q.; Huang, P.-Q. J. Org. Chem. 2004, 69, 6001–6009. doi:10.1021/jo049166z
Return to citation in text: [1] -
Yu, D.-S.; Xu, W.-X.; Liu, L.-X.; Huang, P.-Q. Synlett 2008, 1189–1192. doi:10.1055/s-2008-1072737
Return to citation in text: [1] [2] [3] -
Liu, H.-J.; Yip, J.; Shia, K.-S. Tetrahedron Lett. 1997, 38, 2253–2256. doi:10.1016/s0040-4039(97)00345-6
Return to citation in text: [1] -
Huang, P.-Q.; Wei, B.-G.; Ruan, Y.-P. Synlett 2003, 1663–1667. doi:10.1055/s-2003-40988
Return to citation in text: [1] -
Fukuyama, T.; Frank, R. K.; Jewell, C. F., Jr. J. Am. Chem. Soc. 1980, 102, 2122–2123. doi:10.1021/ja00526a076
Return to citation in text: [1] -
Smith, A. B., III; Wang, Z. J. Org. Chem. 2000, 65, 3738–3753. doi:10.1021/jo991958j
Return to citation in text: [1] -
Tuo, S.; Liu, X.; Huang, P. Chin. J. Chem. 2013, 31, 55–62. doi:10.1002/cjoc.201200904
Return to citation in text: [1] -
Wang, Y.-H.; Ou, W.; Xie, L.-F.; Ye, J.-L.; Huang, P.-Q. Asian J. Org. Chem. 2012, 1, 359–365. doi:10.1002/ajoc.201200113
See for part I of this series.
Return to citation in text: [1]
| 32. |
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
See for an account on the methodology of the regio-and trans-diastereoselective reductive addition of glutarimide. |
| 38. | Zhang, H.-K.; Li, X.; Huang, H.; Huang, P.-Q. Sci. China: Chem. 2011, 54, 737–744. doi:10.1007/s11426-011-4256-4 |
| 45. | Liu, L.-X.; Xiao, K.-J.; Huang, P.-Q. Tetrahedron 2009, 65, 3834–3841. doi:10.1016/j.tet.2009.03.021 |
| 46. | Liu, L.-X.; Ruan, Y.-P.; Guo, Z.-Q.; Huang, P.-Q. J. Org. Chem. 2004, 69, 6001–6009. doi:10.1021/jo049166z |
| 47. | Yu, D.-S.; Xu, W.-X.; Liu, L.-X.; Huang, P.-Q. Synlett 2008, 1189–1192. doi:10.1055/s-2008-1072737 |
| 32. |
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
See for an account on the methodology of the regio-and trans-diastereoselective reductive addition of glutarimide. |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
| 28. | Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015 |
| 29. | O’Mahony, G.; Nieuwenhuyzen, M.; Armstrong, P.; Stevenson, P. J. J. Org. Chem. 2004, 69, 3968–3971. doi:10.1021/jo0497205 |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 50. | Fukuyama, T.; Frank, R. K.; Jewell, C. F., Jr. J. Am. Chem. Soc. 1980, 102, 2122–2123. doi:10.1021/ja00526a076 |
| 51. | Smith, A. B., III; Wang, Z. J. Org. Chem. 2000, 65, 3738–3753. doi:10.1021/jo991958j |
| 49. | Huang, P.-Q.; Wei, B.-G.; Ruan, Y.-P. Synlett 2003, 1663–1667. doi:10.1055/s-2003-40988 |
| 47. | Yu, D.-S.; Xu, W.-X.; Liu, L.-X.; Huang, P.-Q. Synlett 2008, 1189–1192. doi:10.1055/s-2008-1072737 |
| 32. |
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
See for an account on the methodology of the regio-and trans-diastereoselective reductive addition of glutarimide. |
| 48. | Liu, H.-J.; Yip, J.; Shia, K.-S. Tetrahedron Lett. 1997, 38, 2253–2256. doi:10.1016/s0040-4039(97)00345-6 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 28. | Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015 |
| 29. | O’Mahony, G.; Nieuwenhuyzen, M.; Armstrong, P.; Stevenson, P. J. J. Org. Chem. 2004, 69, 3968–3971. doi:10.1021/jo0497205 |
| 29. | O’Mahony, G.; Nieuwenhuyzen, M.; Armstrong, P.; Stevenson, P. J. J. Org. Chem. 2004, 69, 3968–3971. doi:10.1021/jo0497205 |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 28. | Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015 |
| 52. | Tuo, S.; Liu, X.; Huang, P. Chin. J. Chem. 2013, 31, 55–62. doi:10.1002/cjoc.201200904 |
| 53. |
Wang, Y.-H.; Ou, W.; Xie, L.-F.; Ye, J.-L.; Huang, P.-Q. Asian J. Org. Chem. 2012, 1, 359–365. doi:10.1002/ajoc.201200113
See for part I of this series. |
| 47. | Yu, D.-S.; Xu, W.-X.; Liu, L.-X.; Huang, P.-Q. Synlett 2008, 1189–1192. doi:10.1055/s-2008-1072737 |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
| 17. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3315–3325. doi:10.1002/1099-0690(200210)2002:19<3315::AID-EJOC3315>3.0.CO;2-2 |
| 1. | Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556–1575. doi:10.1021/np0580560 |
| 6. | Takada, W.; Sakata, T.; Shimano, S.; Enami, Y.; Mori, N.; Nishida, R.; Kuwahara, Y. J. Chem. Ecol. 2005, 31, 2403–2415. doi:10.1007/s10886-005-7109-9 |
| 13. | Pinho, V. D.; Procter, D. J.; Burtoloso, A. C. B. Org. Lett. 2013, 15, 2434–2437. doi:10.1021/ol400903n |
| 14. | Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w |
| 15. | Sultane, P. R.; Mohite, A. R.; Bhat, R. G. Tetrahedron Lett. 2012, 53, 5856–5858. doi:10.1016/j.tetlet.2012.08.061 |
| 18. | Ni, Y.; Zhao, G.; Ding, Y. J. Chem. Soc., Perkin Trans. 1 2000, 3264–3266. doi:10.1039/b004450o |
| 19. | Barrett, A. G. M.; Damiani, F. J. Org. Chem. 1999, 64, 1410–1411. doi:10.1021/jo9820972 |
| 20. | Cossy, J.; Cases, M.; Gomez Pardo, D. Bull. Soc. Chim. Fr. 1997, 134, 141–144. |
| 21. | Cossy, J.; Cases, M.; Gomez Pardo, D. Synlett 1996, 909–910. doi:10.1055/s-1996-5590 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 4. | Smith, S. Q.; Jones, T. H. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7841–7842. doi:10.1073/pnas.0402599101 |
| 5. | Saporito, R. A.; Garraffo, H. M.; Donnelly, M. A.; Edwards, A. L.; Longino, J. T.; Daly, J. W. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 8045–8050. doi:10.1073/pnas.0402365101 |
| 32. |
Huang, P.-Q. Synlett 2006, 1133–1149. doi:10.1055/s-2006-941565
See for an account on the methodology of the regio-and trans-diastereoselective reductive addition of glutarimide. |
| 2. | Vandendriessche, T.; Abdel-Mottaleb, Y.; Maertens, C.; Cuypers, E.; Sudau, A.; Nubbemeyer, U.; Mebs, D.; Tytgat, J. Toxicon 2008, 51, 334–344. doi:10.1016/j.toxicon.2007.10.011 |
| 3. | Daly, J. W.; Gusovsky, F.; McNeal, E. T.; Secunda, S.; Bell, M.; Creveling, C. R.; Nichizawa, Y.; Overman, L. E.; Sharp, M. J.; Rossignol, D. P. Biochem. Pharmacol. 1990, 40, 315–326. doi:10.1016/0006-2952(90)90694-G |
| 6. | Takada, W.; Sakata, T.; Shimano, S.; Enami, Y.; Mori, N.; Nishida, R.; Kuwahara, Y. J. Chem. Ecol. 2005, 31, 2403–2415. doi:10.1007/s10886-005-7109-9 |
| 1. | Daly, J. W.; Spande, T. F.; Garraffo, H. M. J. Nat. Prod. 2005, 68, 1556–1575. doi:10.1021/np0580560 |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 13. | Pinho, V. D.; Procter, D. J.; Burtoloso, A. C. B. Org. Lett. 2013, 15, 2434–2437. doi:10.1021/ol400903n |
| 14. | Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w |
| 15. | Sultane, P. R.; Mohite, A. R.; Bhat, R. G. Tetrahedron Lett. 2012, 53, 5856–5858. doi:10.1016/j.tetlet.2012.08.061 |
| 18. | Ni, Y.; Zhao, G.; Ding, Y. J. Chem. Soc., Perkin Trans. 1 2000, 3264–3266. doi:10.1039/b004450o |
| 19. | Barrett, A. G. M.; Damiani, F. J. Org. Chem. 1999, 64, 1410–1411. doi:10.1021/jo9820972 |
| 20. | Cossy, J.; Cases, M.; Gomez Pardo, D. Bull. Soc. Chim. Fr. 1997, 134, 141–144. |
| 21. | Cossy, J.; Cases, M.; Gomez Pardo, D. Synlett 1996, 909–910. doi:10.1055/s-1996-5590 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
| 28. | Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015 |
| 29. | O’Mahony, G.; Nieuwenhuyzen, M.; Armstrong, P.; Stevenson, P. J. J. Org. Chem. 2004, 69, 3968–3971. doi:10.1021/jo0497205 |
| 30. | Chen, B.-F.; Tasi, M.-R.; Yang, C.-Y.; Chang, J.-K.; Chang, N.-C. Tetrahedron 2004, 60, 10223–10231. doi:10.1016/j.tet.2004.09.001 |
| 31. | Wang, B.; Fang, F.; Lin, G.-Q. Tetrahedron Lett. 2003, 44, 7981–7984. doi:10.1016/j.tetlet.2003.08.113 |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 13. | Pinho, V. D.; Procter, D. J.; Burtoloso, A. C. B. Org. Lett. 2013, 15, 2434–2437. doi:10.1021/ol400903n |
| 14. | Bernardim, B.; Pinho, V. D.; Burtoloso, A. C. B. J. Org. Chem. 2012, 77, 9926–9931. doi:10.1021/jo301967w |
| 15. | Sultane, P. R.; Mohite, A. R.; Bhat, R. G. Tetrahedron Lett. 2012, 53, 5856–5858. doi:10.1016/j.tetlet.2012.08.061 |
| 16. | Woodin, K. S.; Jamison, T. F. J. Org. Chem. 2007, 72, 7451–7454. doi:10.1021/jo071132e |
| 17. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3315–3325. doi:10.1002/1099-0690(200210)2002:19<3315::AID-EJOC3315>3.0.CO;2-2 |
| 18. | Ni, Y.; Zhao, G.; Ding, Y. J. Chem. Soc., Perkin Trans. 1 2000, 3264–3266. doi:10.1039/b004450o |
| 19. | Barrett, A. G. M.; Damiani, F. J. Org. Chem. 1999, 64, 1410–1411. doi:10.1021/jo9820972 |
| 20. | Cossy, J.; Cases, M.; Gomez Pardo, D. Bull. Soc. Chim. Fr. 1997, 134, 141–144. |
| 21. | Cossy, J.; Cases, M.; Gomez Pardo, D. Synlett 1996, 909–910. doi:10.1055/s-1996-5590 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 23. | Bargar, T. M.; Lett, R. M.; Johnson, P. L.; Hunter, J. E.; Chang, C. P.; Pernich, D. J.; Sabol, M. R.; Dick, M. R. J. Agric. Food Chem. 1995, 43, 1044–1051. doi:10.1021/jf00052a037 |
| 24. | Honda, T.; Hoshi, M.; Kanai, K.; Tsubuki, M. J. Chem. Soc., Perkin Trans. 1 1994, 2091–2101. doi:10.1039/p19940002091 |
| 25. | Honda, T.; Hoshi, M.; Tsubuki, M. Heterocycles 1992, 34, 1515–1518. doi:10.3987/COM-92-6098 |
| 26. | Overman, L. E.; Bell, K. L.; Ito, F. J. Am. Chem. Soc. 1984, 106, 4192–4201. doi:10.1021/ja00327a022 |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 11. | Overman, L. E.; Bell, K. L. J. Am. Chem. Soc. 1981, 103, 1851–1853. doi:10.1021/ja00397a052 |
| 17. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3315–3325. doi:10.1002/1099-0690(200210)2002:19<3315::AID-EJOC3315>3.0.CO;2-2 |
| 10. | Daly, J. W.; Tokuyama, T.; Fujiwara, T.; Higher, R. J.; Karlet, I. L. J. Am. Chem. Soc. 1980, 102, 830–836. doi:10.1021/ja00522a064 |
| 7. | Kibayashi, C. Chem. Pharm. Bull. 2005, 53, 1375–1386. doi:10.1248/cpb.53.1375 |
| 8. | Tang, B. C.; Li, W. D. Chin. J. Org. Chem. 2004, 24, 1151–1158. |
| 9. | Franklin, A. S.; Overman, L. E. Chem. Rev. 1996, 96, 505–522. doi:10.1021/CR950021P |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 12. | Fox, D. N. A.; Lathbury, D.; Mahon, M. F.; Molloy, K. C.; Gallagher, T. J. Am. Chem. Soc. 1991, 113, 2652–2656. doi:10.1021/ja00007a044 |
| 22. | Martin, S. F.; Bur, S. K. Tetrahedron 1999, 55, 8905–8914. doi:10.1016/S0040-4020(99)00452-4 |
| 28. | Wu, H.; Yu, M.; Zhang, Y.; Zhao, G. Chin. J. Chem. 2009, 27, 183–188. doi:10.1002/cjoc.200990015 |
| 33. | Liu, X.-K.; Ye, J.-L.; Ruan, Y.-P.; Li, Y.-X.; Huang, P.-Q. J. Org. Chem. 2013, 78, 35–41. doi:10.1021/jo3014484 |
| 34. | Huo, H.-H.; Xia, X.-E.; Zhang, H.-K.; Huang, P.-Q. J. Org. Chem. 2013, 78, 455–465. doi:10.1021/jo302362b |
| 35. | Liu, X.-K.; Zheng, X.; Ruan, Y.-P.; Ma, J.; Huang, P.-Q. Org. Biomol. Chem. 2012, 10, 1275–1284. doi:10.1039/c1ob06697h |
| 36. | Zhuang, J.-J.; Ye, J.-L.; Zhang, H.-K.; Huang, P.-Q. Tetrahedron 2012, 68, 1750–1755. doi:10.1016/j.tet.2011.12.063 |
| 37. | Liu, X.-K.; Qiu, S.; Xiang, Y.-G.; Ruan, Y.-P.; Zheng, X.; Huang, P.-Q. J. Org. Chem. 2011, 76, 4952–4963. doi:10.1021/jo200600n |
| 38. | Zhang, H.-K.; Li, X.; Huang, H.; Huang, P.-Q. Sci. China: Chem. 2011, 54, 737–744. doi:10.1007/s11426-011-4256-4 |
| 43. | Dimitrov, V.; Panev, S. Tetrahedron: Asymmetry 2000, 11, 1513–1516. doi:10.1016/S0957-4166(00)00099-9 |
| 44. | Tan, C.-H.; Holmes, A. B. Chem.–Eur. J. 2001, 7, 1845–1854. doi:10.1002/1521-3765(20010504)7:9<1845::AID-CHEM1845>3.0.CO;2-2 |
| 39. | Wu, Y. D.; Houk, K. N.; Florez, J.; Trost, B. M. J. Org. Chem. 1991, 56, 3656–3664. doi:10.1021/jo00011a039 |
| 40. | Ashby, E. C.; Laemmle, J. T. Chem. Rev. 1975, 75, 521–546. doi:10.1021/cr60296a005 |
| 41. | Trost, B. M.; Florez, J.; Jebaratnam, D. J. J. Am. Chem. Soc. 1987, 109, 613–615. doi:10.1021/ja00236a067 |
| 42. | Huang, P. Q.; Zhou, W. S. Tetrahedron: Asymmetry 1991, 2, 875–878. doi:10.1016/S0957-4166(00)82199-0 |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
| 27. | Sudau, A.; Münch, W.; Bats, J. W.; Nubbemeyer, U. Eur. J. Org. Chem. 2002, 3304–3314. doi:10.1002/1099-0690(200210)2002:19<3304::AID-EJOC3304>3.0.CO;2-A |
© 2013 Zhang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)