Abstract
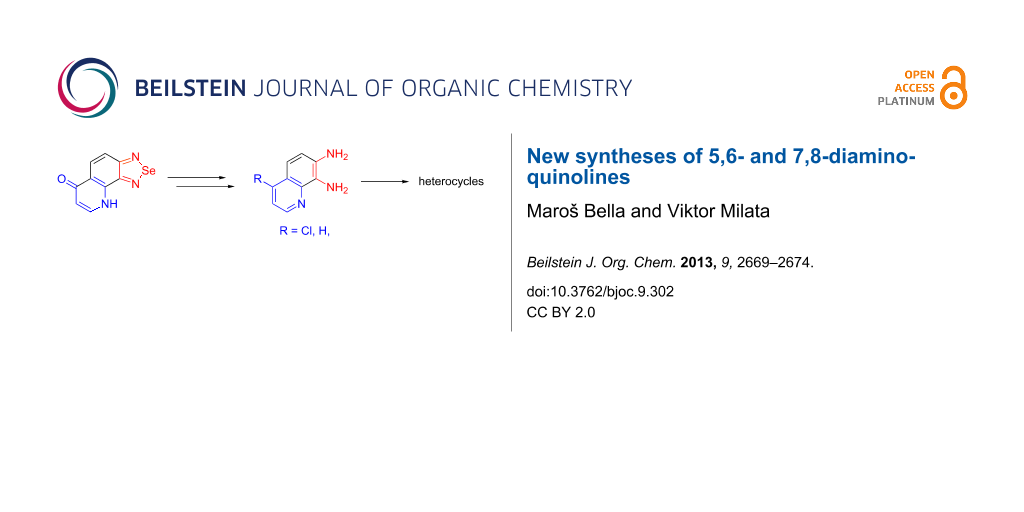
The synthesis of 5,6- and 7,8-diaminoquinoline derivatives starting from angularly annelated selenadiazoloquinolones is presented. Simple chlorination of the pyridone ring followed by reductive deselenation of the 1,2,5-selenadiazole ring afforded novel 4-chloro-o-diaminoquinolines. Dechlorination of 4-chloro-7,8-diaminoquinoline gave 7,8-diaminoquinoline hydrochloride which was successfully employed as starting material in the synthesis of condensed nitrogen heterocycles.

Graphical Abstract
Introduction
o-Diaminoquinolines, in particular 5,6- and 7,8-diaminoquinolines, represent valuable intermediates in the synthesis of nitrogen-containing heterocycles [1-3] including food-borne carcinogens [4].
To date, the known procedures for the synthesis of 7,8-diaminoquinoline have started from 3-nitroaniline or 3-chloroaniline. Initially, 7,8-diaminoquinoline was prepared by Renshaw et al. [5] by coupling 7-aminoquinoline with benzenediazonium chloride followed by the reduction of the resulting azo-dye with SnCl2·2H2O. The second method relies on the Skraup reaction [6] of 3-nitroaniline affording 7-nitroquinoline in a low yield (14%). The latter was reduced with iron in acetic acid to 7-aminoquinoline which was subsequently tosylated, nitrated in position 8 and detosylated to yield 7-amino-8-nitroquinoline. In the final step, the reduction of aminonitroquinoline with SnCl2·2H2O provided 7,8-diaminoquinoline [7]. Another approach is based on the amination of 7-nitroquinoline in position 8 with hydroxylamine under basic conditions. The resulting 8-amino-7-nitroquinoline was reduced with SnCl2·2H2O [8] or hydrazine hydrate on Raney nickel as a catalyst [9] to obtain 7,8-diaminoquinoline. In the last method, the Skraup reaction of 3-chloroaniline led to 7-chloroquinoline which, after nitration, replacement of the chlorine atom by the amino group and catalytic hydrogenation on 5% palladium on charcoal, yielded 7,8-diaminoquinoline [10].
The preparation of 5,6-diaminoquinoline is more effective because the Skraup reaction of 4-nitroaniline produces 6-nitroquinoline as a sole product in moderate yield (47%). The latter, on treatment with hydroxylamine hydrochloride in the presence of KOH [11] followed by the reduction with hydrazine hydrate on Raney nickel, provided 5,6-diaminoquinoline in almost quantitative yield [12].
A disadvantage of the syntheses of o-diaminoquinolines described above is that they require the Skraup reaction, which usually gives low yields and can be violently exothermic [6]. Moreover, in the case of 3-nitro- and 3-chloroaniline, the Skraup reaction affords a mixture of 5- and 7-substituted quinolines, making their separation necessary. To avoid these drawbacks, the readily available angularly annelated selenadiazoloquinolones, prepared as detailed in our previous papers [13,14], were employed as the precursors of o-diaminoquinoline derivatives. The preparation and application of 7,8-diaminoquinoline hydrochloride in the synthesis of nitrogen heterocycles is discussed in datail.
Results and Discussion
In the present approach, selenadiazoloquinolones 1 and 9 represent a masked form of the target o-diaminoquinolines where the o-phenylenediamine moiety is protected as the 2,1,3-benzoselenadiazole skeleton while the pyridine ring can be obtained by transformation of the pyridone core [15]. The synthesis of 7,8-diaminoquinoline (3) starting from selenadiazolo[3,4-h]quinolone 1 is depicted in Scheme 1. In the first step, the aromatization of the pyridone ring by chlorination with POCl3 in DMF, which worked reliably on related azoloquinolones [15], was attempted. However, in the case of selenadiazolo[3,4-h]quinolone 1, only decomposition products were observed. On the other hand, chlorination with neat POCl3 at 90 °C afforded 6-chloroselenadiazoloquinoline 2 in 74% yield after recrystallisation from toluene (Scheme 1). The temperature of the chlorination (90 °C) was found to be crucial, since heating of the reaction mixture under reflux led to a rapid decomposition. The catalytic hydrogenation of 6-chloroderivative 2 using 10% palladium on charcoal or Raney nickel did not afford 7,8-diaminoquinoline (3) in a single step. Finally, the simultaneous deselenation and dechlorination of 6-chloroderivative 2 with zinc in refluxing acetic acid accessed 7,8-diaminoquinoline (3) in 31% yield. Due to the low yield of diaminoquinoline 3 following the treatment with zinc in acetic acid, the reduction of 6-chloroderivative 2 was performed in two successive steps. First, the reductive deselenation with SnCl2·2H2O in concentrated hydrochloric acid afforded 4-chlorodiaminoquinoline 4 in 89% yield. The subsequent catalytic hydrogenation on 10% palladium on charcoal in the absence of base, after filtration of the catalyst and evaporation of methanol, gave 7,8-diaminoquinoline hydrochloride (5) in high yield (Scheme 1). It was notable that the use of sodium hydroxide as a base during the catalytic hydrogenation either on 10% palladium on charcoal or Raney nickel did not lead to diaminoquinoline 3. After alkalisation of the hydrochloride 5 with a sodium hydroxide solution, diaminoquinoline 3 was isolated in good yield (61%). However, 7,8-diaminoquinoline (3) is relatively unstable and decomposes slowly both during isolation or subsequent to it. Hence, the isolation of free diaminoquinoline 3 is practically irreproducible due to this instability. Accordingly, it is far more convenient to employ hydrochloride 5 as the starting material in the subsequent reactions, whether isolated or prepared in situ.
![[1860-5397-9-302-i1]](/bjoc/content/inline/1860-5397-9-302-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 7,8-diaminoquinoline hydrochloride (5).
Scheme 1: Synthesis of 7,8-diaminoquinoline hydrochloride (5).
Next, hydrochloride 5 was applied as the substrate in the synthesis of nitrogen heterocycles (Scheme 2). The treatment of hydrochloride 5 with selenium dioxide in water at room temperature afforded selenadiazoloquinoline 6 in 76% yield (Scheme 2) in comparison with 18% yield obtained by heating of diaminoquinoline 3 with selenium dioxide in dioxane under reflux [9]. To date, pyridoquinoxaline 8 has been prepared in a poor yield by application of the Skraup synthesis to 5-acetylamidoquinoxaline [16]. Notably, 7,8-diaminoquinoline dihydrochloride condensed with glyoxal bisulfite only with the amino group in position 7 and the pyrazine ring was not closed [17]. In the present case, the cyclocondensation of hydrochloride 5 with diimine 7 [18,19] proceeded smoothly at room temperature affording pyridoquinoxaline 8 in high yield (Scheme 2).
![[1860-5397-9-302-i2]](/bjoc/content/inline/1860-5397-9-302-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Application of hydrochloride 5 in the syntheses of nitrogen heterocycles.
Scheme 2: Application of hydrochloride 5 in the syntheses of nitrogen heterocycles.
The same reaction sequence (chlorination and reductive deselenation) was applied to selenadiazolo[3,4-f]quinolone 9 (Scheme 3). The treatment of selenadiazoloquinolone 9 with POCl3 led to 9-chloroselenadiazoloquinoline 10 in high yield. Unlike the reduction of 6-chloroderivative 2, the reductive deselenation of 9-chloroderivative 10 proceeded much more rapidly and 4-chloro-5,6-diaminoquinoline (11) was isolated in 77% yield. However, it should be noted that this reductive deselenation required careful monitoring by TLC (CHCl3/MeOH 100:1, Rf = 0.10) at 10 min intervals in order to detect its accurate termination, because prolongation of the reaction time results in decreased yields.
![[1860-5397-9-302-i3]](/bjoc/content/inline/1860-5397-9-302-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4-chloro-5,6-diaminoquinoline (11).
Scheme 3: Synthesis of 4-chloro-5,6-diaminoquinoline (11).
Conclusion
Angularly annelated selenadiazoloquinolone 1 and 9 were successfully employed as starting materials in the synthesis of 5,6- and 7,8-diaminoquinoline derivatives. 7,8-Diaminoquinoline hydrochloride (5) was prepared in three steps in 57% overall yield starting from selenadiazolo[3,4-h]quinolone 1. Hydrochloride 5 was applied as an important intermediate in the synthesis of nitrogen heterocycles. Hydrochloride 5 affords the advantages of stability in air and simple isolation; it could also be used in further reactions prepared in situ without isolation. In addition, 4-chloro-o-diaminoquinolines 4 and 11 also represent valuable substrates for the preparation of chlorinated nitrogen heterocycles.
Experimental
General. Thin-layer chromatography (TLC) was performed on aluminium plates precoated with 0.2 mm silica gel (25 μm) containing fluorescent indicator 254 nm (Fluka) and stains were visualised by UV light (254 nm or 366 nm). Flash liquid chromatography (FLC) was performed on silica gel [Normasil 60 (43–60 μm)]. Melting points were measured on a Koffler block and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury 300 MHz spectrometer at 25 °C. The operating frequencies were 300 MHz for 1H and 75.5 MHz for 13C nuclei. Chemical shifts (δ) are reported in ppm and coupling constants (J) are given in Hz. Elemental analyses were determined using a Thermo Finnigan Flash EA 1112 instrument.
6-Chloro-[1,2,5]selenadiazolo[3,4-h]quinoline (2). A mixture of selenadiazolo[3,4-h]quinolone 1 (5.0 g, 20.0 mmol) and POCl3 (10 mL, 16.4 g, 0.1 mol) was stirred at 90 °C for 15 min. After the reaction was complete, the mixture was cooled to 0 °C in an ice bath followed by the addition of crushed ice (~45 g) in one portion under stirring. Once the ice was melted, the resulting brown solution was alkalised with a 20% NaOH solution under cooling in the ice bath. The brown precipitate was collected by suction, washed with acetone and dried. Recrystallisation from toluene gave 6-chloroselenadiazoloquinoline 2 (4.0 g, 74%) as golden plates; mp 237–240 °C; 1H NMR (300 MHz, TFA-d) δ 8.08 (d, J = 9.8 Hz, 1H, H-4), 8.17 (d, J = 6.0 Hz, 1H, H-7), 8.30 (d, J = 9.8 Hz, 1H, H-5), 8.86 (d, J = 6.0 Hz, 1H, H-8); 13C NMR (75 MHz, TFA-d) δ 127.1, 128.5, 128.8, 131.1, 136.8, 143.6, 151.9, 158.5, 161.4; anal. calcd for C9H4ClN3Se: C, 40.25; H, 1.50; N, 15.65; found: C, 40.19; H, 1.48; N, 15.70.
4-Chloro-7,8-diaminoquinoline (4). SnCl2·2H2O (4.0 g, 17.7 mmol) was added in small portions to a stirred suspension of 6-chloroselenadiazoloquinoline 2 (1.0 g, 3.7 mmol) in concentrated HCl (33 mL) at room temperature and stirring was continued for 6 h. Next, the reaction mixture was diluted with water (30 mL), the insoluble material was removed by filtration under reduced pressure and the filter cake was washed thoroughly with water (150–200 mL). The filtrate was subsequently alkalised with a 20% NaOH solution under cooling in an ice bath and the resulting yellow suspension was extracted with ethyl acetate (3 × 100 mL). The combined organic phases were dried with Na2SO4, filtered and the solvent evaporated under reduced pressure to afford 4-chlorodiaminoquinoline 4 (0.64 g, 89%) as a yellow solid which was used in the next reaction without further purification; mp 136–139 °C. 1H NMR (300 MHz, DMSO-d6) δ 5.19 (br s, 2H, NH2), 5.25 (br s, 2H, NH2), 7.14 (d, J = 8.7 Hz, 1H, H-6), 7.28 (d, J = 8.7 Hz, 1H, H-5), 7.29 (d, J = 4.6 Hz, 1H, H-3), 8.50 (d, J = 4.6, 1H, H-2); 13C NMR (75 MHz, DMSO-d6) δ 110.9, 116.7, 118.5, 119.4, 127.0, 133.7, 138.3, 140.7, 146.8; anal. calcd for C9H8ClN3: C, 55.83; H, 4.16; N, 21.70; found: C, 55.92; H, 4.20; N, 21.61.
7,8-Diaminoquinoline hydrochloride (5). 10% Pd/C (0.40 g) was added to a solution of 4-chlorodiaminoquinoline 4 (0.64 g, 3.3 mmol) in methanol (35 mL) and the resulting mixture was stirred under H2 atmosphere (balloon) overnight at room temperature. Once the reaction was complete, the catalyst was removed by filtration and the dark red filtrate was evaporated under reduced pressure. CHCl3 (35 mL) was added to the dark violet solid thus obtained and the resulting suspension was stirred for 15 min. Insoluble material was collected by suction, washed with CHCl3 and dried to give hydrochloride 5 (0.55 g, 86%) as a dark violet solid which was used in the next reactions without further purification; mp >230 °C (dec.). 1H NMR (300 MHz, DMSO-d6) δ 7.31 (d, 1H, J = 8.8 Hz, H-6), 7.41 (dd, 1H, J = 8.0, 5.0 Hz, H-3), 7.53 (d, 1H, J = 8.8 Hz, H-5), 8.48 (dd, 1H, J = 8.0, 1.1 Hz, H-4), 8.75 (dd, 1H, J = 5.0, 1.5 Hz, H-2); 13C NMR (75 MHz, DMSO-d6) δ 116.7, 120.7, 121.3, 122.8, 140.3, 145.2, 164.2 (not all 13C carbon signals were observed); anal. calcd for C9H10ClN3: C, 55.25; H, 5.15; N, 21.48, found: C, 55.34; H, 5.18; N, 21.55.
7,8-Diaminoquinoline (3). Method A (alkalisation of hydrochloride 5). A solution of hydrochloride 5 (0.24 g, 1.2 mmol) in water (25 mL) was alkalised with a few drops of 20% NaOH solution. The resulting solution was filtered under reduced pressure and the filtrate was extracted with ethyl acetate (4 × 15 mL). The combined organic phases were dried with Na2SO4, filtered and evaporated under reduced pressure as rapidly as possible to afford diaminoquinoline 3 (0.12 g, 61%) as a dark grey solid which was characterised without further purification; mp 100–102 °C (Ref. [8] mp 102 °C). 1H NMR (300 MHz, DMSO-d6) δ 5.07 (br s, 4H, 2×NH2), 7.02 (d, J = 8.6 Hz, 1H, H-6), 7.05 (d, J = 8.6 Hz, 1H, H-5), 7.11 (dd, J = 8.1, 4.2 Hz, 1H, H-3), 8.00 (dd, J = 8.1, 1.7 Hz, 1H, H-4), 8.59 (dd, J = 4.2, 1.7 Hz, 1H, H-2); 13C NMR (75 MHz, DMSO-d6) δ 115.5, 116.7, 118.7, 121.4, 126.3, 132.6, 135.5, 137.9, 147.3; anal. calcd for C9H9N3: C, 67.90; H, 5.70; N, 26.40; found: C, 68.00; H, 5.65; N, 26.47.
Method B (reduction of 6-chloroselenadiazoloquinoline 2). Powdered Zn (0.58 g, 8.93 mmol) was added to a stirred suspension of 6-chloroselenadiazoloquinoline 2 (0.20 g, 0.74 mmol) in acetic acid and the mixture was heated under reflux for 4 h. After the reaction was complete, the hot mixture was filtered, the filter cake was washed with acetic acid (15 mL) and the filtrate was evaporated under reduced pressure. The resulting orange oily residue was dissolved in water (7 mL) and alkalised with a 10% NaOH solution. The yellow suspension was extracted with ethyl acetate (4 × 15 mL). The combined organic phases were dried with Na2SO4, filtered and evaporated under reduced pressure as rapidly as possible to afford diaminoquinoline 3 (37 mg, 31%) as a dark grey solid which was characterised without further purification; mp 99–101 °C (Ref. [8] mp 102 °C). The 1H and 13C NMR spectral data were in accordance with those of diaminoquinoline 3 prepared by the alkalisation of hydrochloride 5.
[1,2,5]Selenadiazolo[3,4-h]quinoline (6). SeO2 (58.6 mg, 0.53 mmol) dissolved in water (1 mL) was added to a stirred solution of hydrochloride 5 (100 mg, 0.51 mmol) in water (1 mL) and stirring was continued for an additional 15 min at room temperature. Next, the reaction mixture was alkalised with a few drops of 30% NaOH solution and the resulting grey precipitate was collected by suction, washed with water (15 mL) and dried. The mother liquor was extracted with CHCl3 (1 × 20 mL), the organic layer was dried with Na2SO4, filtered and evaporated under reduced pressure. The combined solids obtained after separation by suction and extraction were purified by FLC (silica gel, CHCl3/MeOH 100:1, Rf = 0.31) to give selenadiazoloquinoline 6 (92 mg, 76%) as brownish needles; mp 153–155 °C (Ref. [9] mp 150–151 °C). 1H NMR (300 MHz, CDCl3) δ 7.62 (dd, 1H, J = 8.0, 4.4 Hz, H-7), 7.71 (d, 1H, J = 9.5 Hz, H-4), 7.77 (d, 1H, J = 9.5 Hz, H-5), 8.14 (dd, 1H, J = 8.0, 1.4 Hz, H-6), 9.04 (dd, 1H, J = 4.4, 1.4 Hz, H-8); 13C NMR (75 MHz, CDCl3) δ 123.1, 124.0, 128.6, 130.6, 136.0, 144.4, 150.1, 158.5, 160.7; anal. calcd for C9H5N3Se: C, 46.17; H, 2.15; N, 17.95, found: C, 46.20; H, 2.14; N, 18.00.
Pyrido[2,3-f]quinoxaline (8). Diimine 7 (113 mg, 0.51 mmol) was added to a stirred solution of hydrochloride 5 (100 mg, 0.51 mmol) in MeOH (8 mL) and stirring was continued at room temperature for 1.5 h. The volatiles were evaporated under reduced pressure and the residue was purified by FLC (silica gel, CHCl3/MeOH 100:1, Rf = 0.25) to afford pyridoquinoxaline 8 (75 mg, 81%) as an off-white solid; mp 147–149 °C (Ref. [16] mp 146.5–147.5 °C). 1H NMR (300 MHz, CDCl3) δ 7.69 (dd, 1H, J = 8.1, 4.3 Hz, H-8), 8.03 (d, 1H, J = 9.1 Hz, H-5), 8.08 (d, 1H, J = 9.1 Hz, H-6), 8.30 (dd, 1H, J = 8.1, 1.6 Hz, H-7), 9.01 (d, 1H, J = 1.9 Hz, H-3), 9.10 (d, 1H, J = 1.9 Hz, H-2), 9.21 (dd, 1H, J = 4.3, 1.6 Hz, H-9); 13C NMR (75 MHz, CDCl3) δ 123.6, 128.1, 128.4, 130.1, 136.1, 141.3, 144.4, 144.5, 145.5, 145.7, 150.7; anal. calcd for C11H7N3: C, 72.92; H, 3.89; N, 23.19; found: C, 72.97; H, 3.90; N, 23.17.
9-Chloro-[1,2,5]selenadiazolo[3,4-f]quinoline (10). A mixture of selenadiazolo[3,4-f]quinolone 9 (2.0 g, 8.0 mmol) and POCl3 (4 mL, 6.56 g, 42.8 mmol) was stirred at 90 °C for 3 h. After the reaction was complete, the mixture was cooled to 0 °C in an ice bath followed by the addition of crushed ice (~30 g) in one portion under stirring. Once the ice was melted, the resulting brown solution was stirred for 45 min and subsequently alkalised with a 20% NaOH solution under cooling in the ice bath. The grey–brown precipitate was collected by suction, washed with water and dried. Purification by FLC (silica gel, CHCl3, Rf = 0.21) afforded 9-chloroselenadiazoloquinoline 10 (1.90 g, 89%) as a pale yellow solid; mp 225–226 °C. 1H NMR (300 MHz, TFA-d) δ 8.02 (d, 1H, J = 9.7 Hz, H-4), 8.06 (d, 1H, J = 6.3 Hz, H-8), 8.26 (d, 1H, J = 9.7 Hz, H-5), 8.69 (d, 1H, J = 6.1 Hz, H-7); 13C NMR (75 MHz, TFA-d) δ 125.2, 126.1, 129.9, 134.5, 143.3, 145.8, 155.2, 158.6, 159.7; anal. calcd for C9H4ClN3Se: C, 40.25; H, 1.50; N, 15.65; found: C, 40.21; H, 1.51; N, 15.67.
4-Chloro-5,6-diaminoquinoline (11). 9-Chloroselenadiazoloquinoline 10 (0.50 g, 1.86 mmol) was added in small portions to a stirred suspension of SnCl2·2H2O (1.68 g, 7.44 mmol) in concentrated HCl (15 mL) at room temperature and stirring was continued for 30 min. Next, the reaction mixture was diluted with water (40 mL), the insoluble material was removed by filtration under reduced pressure and the filter cake was washed with water (20 mL). The filtrate was subsequently alkalised with saturated Na2CO3 solution and the resulting yellow suspension was extracted with ethyl acetate (6 × 75 mL). In the first extraction, precipitation of the product occurred between the organic and water phases due to its low solubility in ethyl acetate. In this case, the water phase was separated to leave the precipitate in the organic phase in a separatory funnel. Methanol (15 mL) was added to this suspension to dissolve the precipitated product and the resulting yellow solution was put aside. The water phase was extracted with 5 additional portions of ethyl acetate. The combined organic phases were dried with Na2SO4, filtered and evaporated under reduced pressure to afford 4-chlorodiaminoquinoline 11 (0.28 g, 77%) as a yellow–brown solid which was characterised without further purification; mp > 150 °C (dec.). 1H NMR (300 MHz, DMSO-d6) δ 5.13 (br s, 2H, NH2), 5.23 (br s, 2H, NH2), 7.27 (s, 2H, H-7, H-8), 7.28 (d, 1H, J = 4.5 Hz, H-3), 8.29 (d, 1H, J = 4.5 Hz, H-2); 13C NMR (75 MHz, DMSO-d6) δ 114.8, 118.9, 121.5, 121.6, 125.8, 132.8, 136.8, 144.52, 144.53; anal. calcd for C9H8ClN3: C, 55.83; H, 4.16; N, 21.70, found: C, 55.93; H, 4.12; N, 21.79.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR spectra of compounds 2–6, 8, 10 and 11. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Aleksandrov, A. A.; Dedeneva, A. S.; Vlasova, E. V.; El'chaninov, M. M. Russ. J. Org. Chem. 2011, 47, 120–123. doi:10.1134/S1070428011010155
Return to citation in text: [1] -
Lee, J. H.; Ahn, M. H.; Choi, E. H.; Choo, H.-Y. P.; Han, G. Heterocycles 2006, 70, 571–580. doi:10.3987/COM-06-S(W)25
Return to citation in text: [1] -
Rajitha, B.; Rao, M. K.; Reddy, P. N. Indian J. Chem., Sect. B 2004, 43, 417–419.
Return to citation in text: [1] -
Ziv, J.; Knapp, S.; Rosen, J. D. Synth. Commun. 1988, 18, 973–980. doi:10.1080/00397918808060881
Return to citation in text: [1] -
Renshaw, R. R.; Friedman, H. L.; Gajewski, F. J. J. Am. Chem. Soc. 1939, 61, 3322–3326. doi:10.1021/ja01267a026
Return to citation in text: [1] -
Hudson, A. Skraup/Doebner-von Miller reaction. In Name Reactions in Heterocyclic Chemistry; Li, J. J., Ed.; Wiley & Sons: Hoboken, NJ, 2005; pp 488–494.
Return to citation in text: [1] [2] -
Linsker, F.; Evans, R. L. J. Am. Chem. Soc. 1946, 68, 149–150. doi:10.1021/ja01205a513
Return to citation in text: [1] -
Colonna, M.; Montanari, F. Gazz. Chim. Ital. 1951, 81, 744–756.
Return to citation in text: [1] [2] [3] -
Sharma, K. S.; Kumari, S.; Singh, R. P. Synthesis 1981, 316–318. doi:10.1055/s-1981-29435
Return to citation in text: [1] [2] [3] -
Moores, I. G.; Smalley, R. K.; Suschitzky, H. J. Fluorine Chem. 1982, 20, 573–580. doi:10.1016/S0022-1139(00)82282-4
Return to citation in text: [1] -
Huisgen, R. Justus Liebigs Ann. Chem. 1948, 559, 101–152. doi:10.1002/jlac.19485590204
Return to citation in text: [1] -
Sharma, K. S.; Kumari, S.; Singh, R. P. Indian J. Chem., Sect. B 1981, 20, 744–746.
Return to citation in text: [1] -
Bella, M.; Schultz, M.; Milata, V.; Koňariková, K.; Breza, M. Tetrahedron 2010, 66, 8169–8174. doi:10.1016/j.tet.2010.08.044
Return to citation in text: [1] -
Bella, M.; Schultz, M.; Milata, V. ARKIVOC 2012, No. iv, 242–251. doi:10.3998/ark.5550190.0013.418
Return to citation in text: [1] -
Milata, V.; Ilavský, D.; Leško, J. Collect. Czech. Chem. Commun. 1988, 53, 1068–1077. doi:10.1135/cccc19881068
Return to citation in text: [1] [2] -
Case, F. H.; Brennan, J. A. J. Am. Chem. Soc. 1959, 81, 6297–6301. doi:10.1021/ja01532a046
Return to citation in text: [1] [2] -
Linsker, F.; Evans, R. L. J. Am. Chem. Soc. 1946, 68, 874–876. doi:10.1021/ja01209a051
Return to citation in text: [1] -
Carson, J. F. J. Am. Chem. Soc. 1953, 75, 4337–4338. doi:10.1021/ja01113a507
Return to citation in text: [1] -
Grivas, S.; Tian, W.; Ronne, E.; Lindström, S.; Olsson, K. Acta Chem. Scand. 1993, 47, 521–528. doi:10.3891/acta.chem.scand.47-0521
Return to citation in text: [1]
| 17. | Linsker, F.; Evans, R. L. J. Am. Chem. Soc. 1946, 68, 874–876. doi:10.1021/ja01209a051 |
| 18. | Carson, J. F. J. Am. Chem. Soc. 1953, 75, 4337–4338. doi:10.1021/ja01113a507 |
| 19. | Grivas, S.; Tian, W.; Ronne, E.; Lindström, S.; Olsson, K. Acta Chem. Scand. 1993, 47, 521–528. doi:10.3891/acta.chem.scand.47-0521 |
| 1. | Aleksandrov, A. A.; Dedeneva, A. S.; Vlasova, E. V.; El'chaninov, M. M. Russ. J. Org. Chem. 2011, 47, 120–123. doi:10.1134/S1070428011010155 |
| 2. | Lee, J. H.; Ahn, M. H.; Choi, E. H.; Choo, H.-Y. P.; Han, G. Heterocycles 2006, 70, 571–580. doi:10.3987/COM-06-S(W)25 |
| 3. | Rajitha, B.; Rao, M. K.; Reddy, P. N. Indian J. Chem., Sect. B 2004, 43, 417–419. |
| 7. | Linsker, F.; Evans, R. L. J. Am. Chem. Soc. 1946, 68, 149–150. doi:10.1021/ja01205a513 |
| 9. | Sharma, K. S.; Kumari, S.; Singh, R. P. Synthesis 1981, 316–318. doi:10.1055/s-1981-29435 |
| 6. | Hudson, A. Skraup/Doebner-von Miller reaction. In Name Reactions in Heterocyclic Chemistry; Li, J. J., Ed.; Wiley & Sons: Hoboken, NJ, 2005; pp 488–494. |
| 16. | Case, F. H.; Brennan, J. A. J. Am. Chem. Soc. 1959, 81, 6297–6301. doi:10.1021/ja01532a046 |
| 5. | Renshaw, R. R.; Friedman, H. L.; Gajewski, F. J. J. Am. Chem. Soc. 1939, 61, 3322–3326. doi:10.1021/ja01267a026 |
| 15. | Milata, V.; Ilavský, D.; Leško, J. Collect. Czech. Chem. Commun. 1988, 53, 1068–1077. doi:10.1135/cccc19881068 |
| 4. | Ziv, J.; Knapp, S.; Rosen, J. D. Synth. Commun. 1988, 18, 973–980. doi:10.1080/00397918808060881 |
| 15. | Milata, V.; Ilavský, D.; Leško, J. Collect. Czech. Chem. Commun. 1988, 53, 1068–1077. doi:10.1135/cccc19881068 |
| 11. | Huisgen, R. Justus Liebigs Ann. Chem. 1948, 559, 101–152. doi:10.1002/jlac.19485590204 |
| 6. | Hudson, A. Skraup/Doebner-von Miller reaction. In Name Reactions in Heterocyclic Chemistry; Li, J. J., Ed.; Wiley & Sons: Hoboken, NJ, 2005; pp 488–494. |
| 16. | Case, F. H.; Brennan, J. A. J. Am. Chem. Soc. 1959, 81, 6297–6301. doi:10.1021/ja01532a046 |
| 10. | Moores, I. G.; Smalley, R. K.; Suschitzky, H. J. Fluorine Chem. 1982, 20, 573–580. doi:10.1016/S0022-1139(00)82282-4 |
| 13. | Bella, M.; Schultz, M.; Milata, V.; Koňariková, K.; Breza, M. Tetrahedron 2010, 66, 8169–8174. doi:10.1016/j.tet.2010.08.044 |
| 14. | Bella, M.; Schultz, M.; Milata, V. ARKIVOC 2012, No. iv, 242–251. doi:10.3998/ark.5550190.0013.418 |
| 9. | Sharma, K. S.; Kumari, S.; Singh, R. P. Synthesis 1981, 316–318. doi:10.1055/s-1981-29435 |
| 12. | Sharma, K. S.; Kumari, S.; Singh, R. P. Indian J. Chem., Sect. B 1981, 20, 744–746. |
| 9. | Sharma, K. S.; Kumari, S.; Singh, R. P. Synthesis 1981, 316–318. doi:10.1055/s-1981-29435 |
© 2013 Bella and Milata; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)