Abstract
Density functional theory calculations on mechanisms of the formation of caryolene, a putative biosynthetic precursor to caryol-1(11)-en-10-ol, reveal two mechanisms for caryolene formation: one involves a base-catalyzed deprotonation/reprotonation sequence and tertiary carbocation minimum, whereas the other (with a higher energy barrier) involves intramolecular proton transfer and the generation of a secondary carbocation minimum and a hydrogen-bridged minimum. Both mechanisms are predicted to involve concerted suprafacial/suprafacial [2 + 2] cycloadditions, whose asynchronicity allows them to avoid the constraints of orbital symmetry.

Graphical Abstract
Introduction
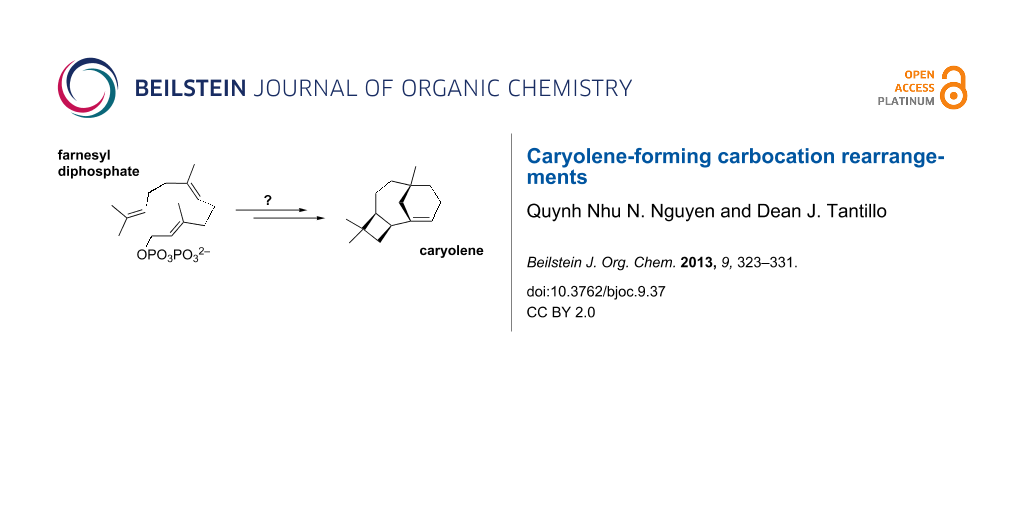
The cytotoxic sesquiterpenol caryol-1(11)-en-10-ol (1, Figure 1) was isolated by Barrow et al. in 1988 during an investigation of antiviral/antitumor compounds from New Zealand marine invertebrates [2]. Similar sesquiterpenoids were also found in Campanella fungi, Streptomyces bacteria, Sinacalia tangutica plants, and Eurypon sponges (Figure 1) [3-6]. The carbon skeleton of 1 is unusual, not only because it contains concatenated 4-, 6-, and 7-membered rings, but also in that it bears a bridgehead double bond. As noted in the original isolation report [2], this type of bridgehead C=C bond is rare for naturally occurring compounds, since it is expected to be associated with significant strain (although it is not technically in violation of Bredt’s Rule) [7]. Intrigued by this structure, we proposed a biosynthetic mechanism for its formation (Scheme 1) and set about putting this proposal to the test using quantum chemical calculations [8].
![[1860-5397-9-37-1]](/bjoc/content/figures/1860-5397-9-37-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Caryol-1(11)-en-10-ol (1) and similar sesquiterpenoids. Note that a different atom numbering was used in the paper describing the isolation of 1 [2].
Figure 1: Caryol-1(11)-en-10-ol (1) and similar sesquiterpenoids. Note that a different atom numbering was us...
![[1860-5397-9-37-i1]](/bjoc/content/inline/1860-5397-9-37-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Initially proposed mechanism for caryolene (caryol-1(11)-en-10-ol, 1) formation. Atom numbers for farnesyl diphosphate are shown. These are used in the mechanistic discussions herein; note that this numbering system differs from that generally used for caryolene and caryolenol.
Scheme 1: Initially proposed mechanism for caryolene (caryol-1(11)-en-10-ol, 1) formation. Atom numbers for f...
Results and Discussion
Structure validation: We first computed 1H and 13C chemical shifts for 1 to assure ourselves that the assigned structure was reasonable [9,10]. Our calculated chemical shifts and the reported data matched well (Figure 2). The mean absolute deviations between computed and experimental chemical shifts were 0.10 ppm for 1H and 1.96 ppm for 13C, and the largest deviations were 0.31 ppm and 4.40 ppm for 1H and 13C, respectively. These values are typical for structures known to be correct [11-16], giving us confidence in the original structural assignment. Interestingly, our calculations also indicate that the bridgehead C=C unit of 1 is not actually associated with much geometric strain [17].
![[1860-5397-9-37-2]](/bjoc/content/figures/1860-5397-9-37-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Computed (top) and experimental (bottom, underlined italics) [2] 1H and 13C chemical shifts for 1 (lowest-energy conformer found); see Experimental for details.
Figure 2: Computed (top) and experimental (bottom, underlined italics) [2] 1H and 13C chemical shifts for 1 (low...
Proposed mechanism: Initially, we proposed the pathway shown in Scheme 1 for caryolene formation, applying the principles derived from previous theoretical studies on terpene-forming carbocation rearrangements [8]. In this mechanism, formation of the C1–C11 bond was expected to result in secondary carbocation B, in analogy to previously characterized pathways to sesquiterpenes containing 11-membered rings [1,8,18-22]. The C2=C3 π-bond was then expected to attack C10 to form the 4-membered ring (see C), in analogy to previously proposed mechanisms for caryophyllene formation [21,23]. An intramolecular proton transfer from the C15 methyl group to the nearby C6=C7 π-bond could then generate D. Related intramolecular proton transfers have been described [24-33]. Attack of the resulting C3=C15 π-bond onto C7 would complete the carbon skeleton of 1, leaving a bridgehead carbocation [34], whose deprotonation would lead to caryolene, the putative biosynthetic precursor to 1. Despite the apparent reasonability of this proposed mechanism, our quantum chemical calculations indicated that the pathway as formulated in Scheme 1 is not energetically viable (see below).
Computed mechanism: The first deviation from the proposed mechanism in Scheme 1 was encountered in the very first step involving carbocations. We were unable to locate a minimum for B in a productive conformation, despite the fact that alternative conformers of this secondary carbocation had been found to be involved in pathways to pentalenene and presilphiperfolanol [18-22]. Instead, a transition-state structure connected directly (by an IRC; see Experimental for details) to farnesyl cation A and cyclobutylcarbinyl cation C was located: TS-AC (Figure 3 and Figure 4). This process bypasses the generation of a secondary carbocation as a minimum [22], and overall corresponds to a formally orbital-symmetry-forbidden [π2S + π2S] cycloaddition [35,36]. Although this process is predicted to be concerted, the bond-forming events occur asynchronously [21,37,38] (note that the C1–C11 distance in TS-AC is approximately 1 Å shorter than the C2–C10 distance, indicating that C1–C11 bond formation leads C2–C10 bond formation) and at no point along the reaction coordinate is there significant cyclic delocalization of the sort that would be associated with a forbidden reaction. As described for other carbocation reactions [39,40], the constraints of orbital symmetry appear to have been circumvented. Interestingly, there is a shoulder (i.e., a sharp downturn) on the reaction coordinate in the vicinity of structures resembling B (Figure 4).
![[1860-5397-9-37-3]](/bjoc/content/figures/1860-5397-9-37-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Computed minima and transition-state structure involved in the single-step conversion of A to C. Relative energies shown (kcal/mol) were calculated at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level.
Figure 3: Computed minima and transition-state structure involved in the single-step conversion of A to C. Re...
![[1860-5397-9-37-4]](/bjoc/content/figures/1860-5397-9-37-4.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 4: IRC from TS-AC toward C. Relative energies were calculated at the B3LYP/6-31+G(d,p) level.
Figure 4: IRC from TS-AC toward C. Relative energies were calculated at the B3LYP/6-31+G(d,p) level.
In the structure of C (Figure 3), C3 is quite close to C6 (only 2.06 Å away), indicating that this structure is perhaps best described as a hybrid of the tertiary cation C and a resonance structure with two 4-membered rings [22]. We performed a conformational search for C to assess whether other conformers lacking this close contact were possible, but all starting geometries examined in which the two methyl groups pointed to the same side of the ring as the first-formed cyclobutane, converged to the structure of C shown in Figure 3 [41].
Locating a pathway for the conversion of C to D (Scheme 1) also proved difficult. We expected proton transfer from the C15 methyl group to C6 of the C6=C7 π-bond to result in tertiary carbocation D. Surprisingly, only a transition-state structure for migration of the proton to C7 instead of C6, generating the secondary cation F (Scheme 2, left and Figure 5), was found. Attempts to independently locate D led instead to G (Figure 5), a nonclassical carbocation [42-46], or back to C [47]. Carbocation G contains a hydrogen bridge between C6 and C7, which also appears to interact with the nearby C3=C15 π-bond. If the interaction with the C3=C15 π-bond were stronger, this structure could be regarded as a “proton sandwich” [20,22,48]. An interesting question thus emerges about the nature of this structure: does the bridging hydrogen have hydride character (as expected for a structure resembling a transition-state structure for a 1,2-hydrogen shift) or proton character (as expected for a “proton sandwich”)? This issue was addressed through calculations of 1H chemical shifts, which predicted a chemical shift of +4.0 ppm for the bridging hydrogen in G. Although this shift is not as far downfield as that predicted for structures with bridging protons (e.g., the predicted shift for the migrating proton in TS-CF is +9.1 ppm and the predicted shifts for symmetric “proton sandwiches” are around +13 ppm) [20], it is well downfield of shifts predicted for hydrides involved in three-center two-electron bonding arrays (e.g., see Supporting Information File 1 for a model transition-state structure for a 1,2-hydride shift with a predicted chemical shift of +1.9 ppm) [49-51] and approximately 2 ppm downfield of its value when merely hyperconjugated in C (+2.6 ppm; computed partial charges paint a similar, but less clear-cut picture; see Supporting Information File 1 for details).
![[1860-5397-9-37-i2]](/bjoc/content/inline/1860-5397-9-37-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Alternative mechanisms for caryolene formation.
Scheme 2: Alternative mechanisms for caryolene formation.
![[1860-5397-9-37-5]](/bjoc/content/figures/1860-5397-9-37-5.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 5: Computed pathway for the conversion of C to E. Relative energies shown (kcal/mol) were calculated at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level.
Figure 5: Computed pathway for the conversion of C to E. Relative energies shown (kcal/mol) were calculated a...
Transition-state structures for the formation of G from F and E from G were also found (Scheme 2 and Figure 5). The former, TS-FG, resembles a transition-state structure for a typical 1,2-hydride shift, but the predicted chemical shift of the bridging hydrogen in this structure is +4.7 ppm. The developing close contact with the C3=C15 π-bond (the H---C15 distance is only 2.32 Å in TS-FG) also brings this transition-state structure closer to the realm of proton sandwiches. TS-GE looks very similar, but with different longer and shorter H---C partial bonds and a longer H---C15 distance (2.71 Å, consistent with its lower predicted chemical shift, +3.8 ppm). The TS-FG/G/TS-GE energy surface is rather flat (all three structures are predicted to be within 0.5 kcal/mol of each other; Figure 5). The G-to-E reaction involves concerted but asynchronous shifting of the bridging hydrogen toward C6 and subsequent ring closure (C7–C15 bond formation). As illustrated in the IRC plot shown in Figure 6, these two events are essentially separate, with structures part way along the pathway to resembling D. The two events are separated by a conformational reorganization in which some C–C bonds twist and release some strain (e.g., along the C5–C6 bond) while orienting the formally empty p-orbital on C7 toward the C3=C15 π-bond. Although the IRC calculation stopped while C15 and C7 were still 3.12 Å away, optimization of the final point led to E. Thus, a complete pathway to E was found, but this pathway differs in several ways from the pathway we expected to find.
![[1860-5397-9-37-6]](/bjoc/content/figures/1860-5397-9-37-6.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 6: IRC from TS-GE toward E. Relative energies were calculated at the B3LYP/6-31+G(d,p) level. Selected dihedral angles are shown in degrees.
Figure 6: IRC from TS-GE toward E. Relative energies were calculated at the B3LYP/6-31+G(d,p) level. Selected...
Alternative mechanism: The pathway just described represents the inherent reactivity of the carbocations involved in the formation of E, but how might the mechanism change if we allowed for an enzymatic base to be involved? Would the same unusual sequence of events associated with the intramolecular proton transfer persist? Would a lower energetic pathway present itself? No changes to the formation of C were predicted in the presence of an ammonia molecule (a simple model base [52-55]), but a stepwise proton-transfer process was found in which the C15 methyl group was first deprotonated to form an ion·molecule complex (2·NH4+ in Figure 7; 2 is a caryophyllene) and then C6 was protonated to form D (Scheme 2, right). This surprisingly elusive tertiary carbocation appears to be stabilized in the presence of a C–H hydrogen-bond acceptor that interacts with its hyperconjugated hydrogen [52-56]. Here a tertiary carbocation requires selective stabilization in order to exist as a minimum; although it is inherently lower in energy than the secondary cation F (on the basis of single-point calculations without ammonia present), without the intermolecular C–H···X interaction described, there is no barrier for its conversion to E (removal of the ammonia molecule from D·NH3 and reoptimization led to structure E) [57]. Still, even though tertiary carbocation D is predicted to exist as a minimum in the presence of a suitable base at the B3LYP/6-31+G(d,p) level, its conversion to E is predicted to be barrierless at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level (Figure 7 and Figure 8). Thus, this ostensibly normal tertiary carbocation lacks the kinetic stability generally associated with the presence of three alkyl groups. Although cation D can be formed by protonation of an alkene as shown, such a scenario would likely require a separate base and acid positioned on opposite sides of the hydrocarbon substrate due to the steric congestion at its core (note the position of NH3 throughout Figure 7), a scenario that could be probed by deuterium labeling of farnesyl diphosphate if a suitable caryolene synthase were isolated.
![[1860-5397-9-37-7]](/bjoc/content/figures/1860-5397-9-37-7.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 7: Computed pathway for the conversion of C to E in the presence of ammonia. Relative energies shown (kcal/mol) were calculated at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level. Here energies are relative to that for a complex of A and NH3.
Figure 7: Computed pathway for the conversion of C to E in the presence of ammonia. Relative energies shown (...
![[1860-5397-9-37-8]](/bjoc/content/figures/1860-5397-9-37-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Predicted energetics for the conversion of A to E in the absence (blue) and presence (auburn) of ammonia. Relative energies shown (kcal/mol) were calculated at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level. Energies for the pathway with ammonia are relative to those for an A·NH3 complex.
Figure 8: Predicted energetics for the conversion of A to E in the absence (blue) and presence (auburn) of am...
The energetics for both pathways shown in Scheme 2 are summarized in Figure 8. The ammonia-free pathway has a substantial barrier, approximately 25 kcal/mol, after formation of C, but the ammonia-assisted pathway does not. In fact, after a small barrier for the formation of C, the potential energy surface for the deprotonation/reprotonation pathway is rather flat, indicating that once C is formed, transformation to E should be facile, provided that the architecture of the active site supports deprotonation/reprotonation.
Conclusion
Which pathway to caryolene is more likely? On the basis of our computed energetics (Figure 8), we favor a mechanism for caryolene formation that involves a concerted but asynchronous [2 + 2] cycloaddition, deprotonation by an enzyme active-site base (as yet, with identity unknown [55,58-64]), and concerted but asynchronous reprotonation/cyclization (Scheme 2, right). However, several interesting structures with unusual bonding arrays are encountered along the base-free pathway.
Experimental
All calculations were carried out with Gaussian 09 [65]. Geometry optimizations, frequency calculations and intrinsic reaction coordinate (IRC) [66,67] calculations were first carried out with B3LYP/6-31G(d) [68-72]. For IRC calculations, force constants were recalculated after every three points or at every point in the event of prematurely terminated jobs. All molecules were then subjected to optimization and frequency calculations at the B3LYP/6-31+G(d,p) level of theory [8,68-72]. Single-point energies were also calculated at the mPW1PW91/6-31+G(d,p) level [73,74] for comparison, since B3LYP energies are generally unreliable when comparing cyclic and acyclic isomers that differ in the number of σ- and π-bonds [8,74,75]. Chemical shifts (1H and 13C) in chloroform (treated with the SMD solvation model [76]) for selected structures were calculated by using mPW1PW91/6-311+G(2d,p) [11-13,73,74]. Computed scaling factors (slope = −1.0823 for 1H and −1.0448 for 13C; intercept = 31.8486 for 1H and 186.0596 for 13C) were used to convert computed isotropic values into chemical shifts [11-13].
Supporting Information
| Supporting Information File 1: Coordinates and energies for all computed structures, IRC plots, additional computational details and full Gaussian citation. | ||
| Format: PDF | Size: 954.2 KB | Download |
References
-
Zu, L.; Xu, M.; Lodewyk, M. W.; Cane, D. E.; Peters, R. J.; Tantillo, D. J. J. Am. Chem. Soc. 2012, 134, 11369–11371. doi:10.1021/ja3043245
Return to citation in text: [1] -
Barrow, C. J.; Blunt, J. W.; Munro, M. H. G. Aust. J. Chem. 1988, 41, 1755–1761. doi:10.1071/CH9881755
Return to citation in text: [1] [2] [3] [4] -
Fraga, B. M. Nat. Prod. Rep. 2012, 29, 1334–1366. doi:10.1039/c2np20074k
Return to citation in text: [1] -
Zhu, Y.; Zhao, Y.; Huang, G.-D.; Wu, W.-S. Helv. Chim. Acta 2008, 91, 1894–1901. doi:10.1002/hlca.200890203
Return to citation in text: [1] -
Yang, Z.; Yang, Y.; Yang, X.; Zhang, Y.; Zhao, L.; Xu, L.; Ding, Z. Chem. Pharm. Bull. 2011, 59, 1430–1433. doi:10.1248/cpb.59.1430
Return to citation in text: [1] -
Liu, R.; Zhou, Z.-Y.; Xu, D.; Wang, F.; Liu, J.-K. Helv. Chim. Acta 2009, 92, 375–378. doi:10.1002/hlca.200800298
Return to citation in text: [1] -
Bredt, J. Justus Liebigs Ann. Chem. 1924, 437, 1–13. doi:10.1002/jlac.19244370102
Return to citation in text: [1] -
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements.
Return to citation in text: [1] [2] [3] [4] [5] -
Suyama, T. L.; Gerwick, W. H.; McPhail, K. L. Bioorg. Med. Chem. 2011, 19, 6675–6701. doi:10.1016/j.bmc.2011.06.011
Leading references on misassigned natural product structures.
Return to citation in text: [1] -
Nicolaou, K. C.; Snyder, S. A. Angew. Chem., Int. Ed. 2005, 44, 1012–1044. doi:10.1002/anie.200460864
Leading references on misassigned natural product structures.
Return to citation in text: [1] -
Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v
And references therein.
Return to citation in text: [1] [2] [3] -
Jain, R.; Bally, T.; Rablen, P. R. J. Org. Chem. 2009, 74, 4017–4023. doi:10.1021/jo900482q
Return to citation in text: [1] [2] [3] -
CHESHIRE, Chemical Shift Repository with Coupling Constants Added Too. http://cheshirenmr.info.
Return to citation in text: [1] [2] [3] -
Lodewyk, M. W.; Tantillo, D. J. J. Nat. Prod. 2011, 74, 1339–1343. doi:10.1021/np2000446
Return to citation in text: [1] -
Quasdorf, K. W.; Huters, A. D.; Lodewyk, M. W.; Tantillo, D. J.; Garg, N. K. J. Am. Chem. Soc. 2012, 134, 1396–1399. doi:10.1021/ja210837b
Return to citation in text: [1] -
Lodewyk, M. W.; Soldi, C.; Jones, P. B.; Olmstead, M. M.; Rita, J.; Shaw, J. T.; Tantillo, D. J. J. Am. Chem. Soc. 2012, 134, 18550–18553. doi:10.1021/ja3089394
Return to citation in text: [1] -
We computed the geometry of hydrogenated 1, and the carbon skeleton did not relax much upon hydrogenation (see Supporting Information File 1).
Return to citation in text: [1] -
Gutta, P.; Tantillo, D. J. J. Am. Chem. Soc. 2006, 128, 6172–6179. doi:10.1021/ja058031n
Return to citation in text: [1] [2] -
Wang, S. C.; Tantillo, D. J. Org. Lett. 2008, 10, 4827–4830. doi:10.1021/ol801898v
Return to citation in text: [1] [2] -
Gutta, P.; Tantillo, D. J. Angew. Chem., Int. Ed. 2005, 44, 2719–2723. doi:10.1002/anie.200461915
Return to citation in text: [1] [2] [3] [4] -
Tantillo, D. J. J. Phys. Org. Chem. 2008, 21, 561–570. doi:10.1002/poc.1320
Return to citation in text: [1] [2] [3] [4] -
Tantillo, D. J. Chem. Soc. Rev. 2010, 39, 2847–2854. doi:10.1039/b917107j
Return to citation in text: [1] [2] [3] [4] [5] -
Croteau, R.; Gundy, A. Arch. Biochem. Biophys. 1984, 233, 838–841. doi:10.1016/0003-9861(84)90513-7
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2011, 133, 18249–18256. doi:10.1021/ja2055929
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. Nat. Chem. 2009, 1, 384–389. doi:10.1038/nchem.287
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. Org. Lett. 2006, 8, 4601–4604. doi:10.1021/ol061884f
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2009, 7, 4101–4109. doi:10.1039/b908738a
Return to citation in text: [1] -
Williams, C. D.; Carroll, B. J.; Jin, Q.; Rithner, C. D.; Lenger, S. R.; Floss, G. H.; Coates, R. M.; Williams, R. M.; Croteau, R. Chem. Biol. 2000, 7, 969–977. doi:10.1016/S1074-5521(00)00046-6
Return to citation in text: [1] -
Allemann, R. K.; Young, N. J.; Ma, S.; Truhlar, D. G.; Gao, J. J. Am. Chem. Soc. 2007, 129, 13008–13013. doi:10.1021/ja0722067
Return to citation in text: [1] -
Deligeorgopoulou, A.; Taylor, S. E.; Forcat, S.; Allemann, R. K. Chem. Commun. 2003, 2162–2163. doi:10.1039/b306867f
Return to citation in text: [1] -
Cane, D. E.; Prabhakaran, P. C.; Oliver, J. S.; McIlwaine, D. B. J. Am. Chem. Soc. 1990, 112, 3209–3210. doi:10.1021/ja00164a051
Return to citation in text: [1] -
Miller, D. J.; Gao, J.; Truhlar, D. G.; Young, N. J.; Gonzalez, V.; Allemann, R. K. Org. Biomol. Chem. 2008, 6, 2346–2354. doi:10.1039/b804198a
Return to citation in text: [1] -
Surendra, K.; Corey, E. J. J. Am. Chem. Soc. 2008, 130, 8865–8869. doi:10.1021/ja802730a
Return to citation in text: [1] -
Abboud, J.-L. M.; Herreros, M.; Notario, R.; Lomas, J. S.; Mareda, J.; Müller, P.; Rossier, J.-C. J. Org. Chem. 1999, 64, 6401–6410. doi:10.1021/jo990724x
Leading references on bridgehead carbocations.
Return to citation in text: [1] -
Hoffmann, R.; Woodward, R. B. Acc. Chem. Res. 1968, 1, 17–22. doi:10.1021/ar50001a003
Return to citation in text: [1] -
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Return to citation in text: [1] -
Williams, A. Concerted Organic and Bio-Organic Mechanisms; CRC Press: Boca Raton, FL, USA, 2000.
Return to citation in text: [1] -
Dewar, M. J. S. J. Am. Chem. Soc. 1984, 106, 209–219. doi:10.1021/ja00313a042
Return to citation in text: [1] -
Nouri, D. H.; Tantillo, D. J. Tetrahedron 2008, 64, 5672–5679. doi:10.1016/j.tet.2008.04.028
Return to citation in text: [1] -
Sio, V.; Harrison, J. G.; Tantillo, D. J. Tetrahedron 2012, 53, 6919–6922. doi:10.1016/j.tetlet.2012.10.019
Return to citation in text: [1] -
Geometries in which the two methyl groups pointed in opposite directions were also examined, but the resulting structures were not productive for caryolene formation; see Supporting Information File 1 for details.
Return to citation in text: [1] -
Grob, C. A. Acc. Chem. Res. 1983, 16, 426–431. doi:10.1021/ar00096a001
Return to citation in text: [1] -
Brown, H. C. Acc. Chem. Res. 1983, 16, 432–440. doi:10.1021/ar00096a002
Return to citation in text: [1] -
Olah, G. A.; Prakash, G. K. S.; Saunders, M. Acc. Chem. Res. 1983, 16, 440–448. doi:10.1021/ar00096a003
Return to citation in text: [1] -
Walling, C. Acc. Chem. Res. 1983, 16, 448–454. doi:10.1021/ar00096a004
Return to citation in text: [1] -
Brown, H. C. The Nonclassical Ion Problem; Plenum Press: New York, NY, 1977.
With comments by P. v. R. Schleyer.
Return to citation in text: [1] -
D-type structures arising from alternative conformers of C were examined, but these correspond to nonproductive stereoisomers; see Supporting Information File 1 for details.
Return to citation in text: [1] -
Ponec, R.; Bultinck, P.; Gutta, P.; Tantillo, D. J. J. Phys. Chem. A 2006, 110, 3785–3789. doi:10.1021/jp0568430
Return to citation in text: [1] -
McMurry, J. E.; Lectka, T. Acc. Chem. Res. 1992, 25, 47–53. doi:10.1021/ar00013a007
Return to citation in text: [1] -
Ponec, R.; Yuzhakov, G.; Tantillo, D. J. J. Org. Chem. 2004, 69, 2992–2996. doi:10.1021/jo035506p
Return to citation in text: [1] -
Tantillo, D. J.; Hoffmann, R. J. Am. Chem. Soc. 2003, 125, 4042–4043. doi:10.1021/ja021394s
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. J. Org. Chem. 2007, 72, 8877–8881. doi:10.1021/jo701526x
Return to citation in text: [1] [2] -
Lodewyk, M. W.; Gutta, P.; Tantillo, D. J. J. Org. Chem. 2008, 73, 6570–6579. doi:10.1021/jo800868r
Return to citation in text: [1] [2] -
Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2010, 132, 5375–5386. doi:10.1021/ja9084786
Return to citation in text: [1] [2] -
Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2010, 8, 4589–4600. doi:10.1039/c0ob00167h
Return to citation in text: [1] [2] [3] -
Nishio, M. J. Mol. Struct. 2012, 1018, 2–7. doi:10.1016/j.molstruc.2012.03.012
See for leading references on C-H···X interactions.
Return to citation in text: [1] -
Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2009, 131, 7999–8015. doi:10.1021/ja9005332
See for the description of a related scenario.
Return to citation in text: [1] -
Roy, A.; Roberts, F. G.; Wilderman, P. R.; Zhou, K.; Peters, R. J.; Coates, R. M. J. Am. Chem. Soc. 2007, 129, 12453–12460. doi:10.1021/ja072447e
Discussions of pyrophosphate as a possible active site base in terpene synthases.
Return to citation in text: [1] -
Greenhagen, B. T.; O’Maille, P. E.; Noel, J. P.; Chappell, J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9826–9831. doi:10.1073/pnas.0601605103
Return to citation in text: [1] -
Shishova, E. Y.; Di Costanzo, L.; Cane, D. E.; Christianson, D. W. Biochemistry 2007, 46, 1941–1951. doi:10.1021/bi0622524
Return to citation in text: [1] -
Peters, R. J.; Croteau, R. B. Arch. Biochem. Biophys. 2003, 417, 203–211. doi:10.1016/S0003-9861(03)00347-3
Return to citation in text: [1] -
Garms, S.; Chen, F.; Boland, W.; Gershenzon, J.; Köllner, T. G. Phytochemistry 2012, 75, 6–13. doi:10.1016/j.phytochem.2011.12.009
Return to citation in text: [1] -
Zhou, K.; Peters, R. J. Chem. Commun. 2011, 47, 4074–4080. doi:10.1039/c0cc02960b
Return to citation in text: [1] -
Miller, D. J.; Allemann, R. K. Nat. Prod. Rep. 2012, 29, 60–71. doi:10.1039/c1np00060h
Return to citation in text: [1] -
Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2004.
Return to citation in text: [1] -
Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1990, 94, 5523–5527. doi:10.1021/j100377a021
Return to citation in text: [1] -
Fukui, K. Acc. Chem. Res. 1981, 14, 363–368. doi:10.1021/ar00072a001
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304
Return to citation in text: [1] [2] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] [2] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] [2] -
Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001
Return to citation in text: [1] [2] -
Tirado-Rives, J.; Jorgensen, W. L. J. Chem. Theory Comput. 2008, 4, 297–306. doi:10.1021/ct700248k
Return to citation in text: [1] [2] -
Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675. doi:10.1063/1.475428
Return to citation in text: [1] [2] -
Matsuda, S. P. T.; Wilson, W. K.; Xiong, Q. Org. Biomol. Chem. 2006, 4, 530–543. doi:10.1039/b513599k
Return to citation in text: [1] [2] [3] -
Pieniazek, S. N.; Clemente, F. R.; Houk, K. N. Angew. Chem., Int. Ed. 2008, 47, 7746–7749. doi:10.1002/anie.200801843
Return to citation in text: [1] -
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n
Return to citation in text: [1]
| 52. | Hong, Y. J.; Tantillo, D. J. J. Org. Chem. 2007, 72, 8877–8881. doi:10.1021/jo701526x |
| 53. | Lodewyk, M. W.; Gutta, P.; Tantillo, D. J. J. Org. Chem. 2008, 73, 6570–6579. doi:10.1021/jo800868r |
| 54. | Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2010, 132, 5375–5386. doi:10.1021/ja9084786 |
| 55. | Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2010, 8, 4589–4600. doi:10.1039/c0ob00167h |
| 52. | Hong, Y. J.; Tantillo, D. J. J. Org. Chem. 2007, 72, 8877–8881. doi:10.1021/jo701526x |
| 53. | Lodewyk, M. W.; Gutta, P.; Tantillo, D. J. J. Org. Chem. 2008, 73, 6570–6579. doi:10.1021/jo800868r |
| 54. | Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2010, 132, 5375–5386. doi:10.1021/ja9084786 |
| 55. | Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2010, 8, 4589–4600. doi:10.1039/c0ob00167h |
| 56. |
Nishio, M. J. Mol. Struct. 2012, 1018, 2–7. doi:10.1016/j.molstruc.2012.03.012
See for leading references on C-H···X interactions. |
| 57. |
Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2009, 131, 7999–8015. doi:10.1021/ja9005332
See for the description of a related scenario. |
| 2. | Barrow, C. J.; Blunt, J. W.; Munro, M. H. G. Aust. J. Chem. 1988, 41, 1755–1761. doi:10.1071/CH9881755 |
| 8. |
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements. |
| 34. |
Abboud, J.-L. M.; Herreros, M.; Notario, R.; Lomas, J. S.; Mareda, J.; Müller, P.; Rossier, J.-C. J. Org. Chem. 1999, 64, 6401–6410. doi:10.1021/jo990724x
Leading references on bridgehead carbocations. |
| 8. |
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements. |
| 74. | Matsuda, S. P. T.; Wilson, W. K.; Xiong, Q. Org. Biomol. Chem. 2006, 4, 530–543. doi:10.1039/b513599k |
| 75. | Pieniazek, S. N.; Clemente, F. R.; Houk, K. N. Angew. Chem., Int. Ed. 2008, 47, 7746–7749. doi:10.1002/anie.200801843 |
| 7. | Bredt, J. Justus Liebigs Ann. Chem. 1924, 437, 1–13. doi:10.1002/jlac.19244370102 |
| 18. | Gutta, P.; Tantillo, D. J. J. Am. Chem. Soc. 2006, 128, 6172–6179. doi:10.1021/ja058031n |
| 19. | Wang, S. C.; Tantillo, D. J. Org. Lett. 2008, 10, 4827–4830. doi:10.1021/ol801898v |
| 20. | Gutta, P.; Tantillo, D. J. Angew. Chem., Int. Ed. 2005, 44, 2719–2723. doi:10.1002/anie.200461915 |
| 21. | Tantillo, D. J. J. Phys. Org. Chem. 2008, 21, 561–570. doi:10.1002/poc.1320 |
| 22. | Tantillo, D. J. Chem. Soc. Rev. 2010, 39, 2847–2854. doi:10.1039/b917107j |
| 76. | Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378–6396. doi:10.1021/jp810292n |
| 2. | Barrow, C. J.; Blunt, J. W.; Munro, M. H. G. Aust. J. Chem. 1988, 41, 1755–1761. doi:10.1071/CH9881755 |
| 21. | Tantillo, D. J. J. Phys. Org. Chem. 2008, 21, 561–570. doi:10.1002/poc.1320 |
| 23. | Croteau, R.; Gundy, A. Arch. Biochem. Biophys. 1984, 233, 838–841. doi:10.1016/0003-9861(84)90513-7 |
| 8. |
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements. |
| 68. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 69. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 70. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 71. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 72. | Tirado-Rives, J.; Jorgensen, W. L. J. Chem. Theory Comput. 2008, 4, 297–306. doi:10.1021/ct700248k |
| 3. | Fraga, B. M. Nat. Prod. Rep. 2012, 29, 1334–1366. doi:10.1039/c2np20074k |
| 4. | Zhu, Y.; Zhao, Y.; Huang, G.-D.; Wu, W.-S. Helv. Chim. Acta 2008, 91, 1894–1901. doi:10.1002/hlca.200890203 |
| 5. | Yang, Z.; Yang, Y.; Yang, X.; Zhang, Y.; Zhao, L.; Xu, L.; Ding, Z. Chem. Pharm. Bull. 2011, 59, 1430–1433. doi:10.1248/cpb.59.1430 |
| 6. | Liu, R.; Zhou, Z.-Y.; Xu, D.; Wang, F.; Liu, J.-K. Helv. Chim. Acta 2009, 92, 375–378. doi:10.1002/hlca.200800298 |
| 24. | Hong, Y. J.; Tantillo, D. J. J. Am. Chem. Soc. 2011, 133, 18249–18256. doi:10.1021/ja2055929 |
| 25. | Hong, Y. J.; Tantillo, D. J. Nat. Chem. 2009, 1, 384–389. doi:10.1038/nchem.287 |
| 26. | Hong, Y. J.; Tantillo, D. J. Org. Lett. 2006, 8, 4601–4604. doi:10.1021/ol061884f |
| 27. | Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2009, 7, 4101–4109. doi:10.1039/b908738a |
| 28. | Williams, C. D.; Carroll, B. J.; Jin, Q.; Rithner, C. D.; Lenger, S. R.; Floss, G. H.; Coates, R. M.; Williams, R. M.; Croteau, R. Chem. Biol. 2000, 7, 969–977. doi:10.1016/S1074-5521(00)00046-6 |
| 29. | Allemann, R. K.; Young, N. J.; Ma, S.; Truhlar, D. G.; Gao, J. J. Am. Chem. Soc. 2007, 129, 13008–13013. doi:10.1021/ja0722067 |
| 30. | Deligeorgopoulou, A.; Taylor, S. E.; Forcat, S.; Allemann, R. K. Chem. Commun. 2003, 2162–2163. doi:10.1039/b306867f |
| 31. | Cane, D. E.; Prabhakaran, P. C.; Oliver, J. S.; McIlwaine, D. B. J. Am. Chem. Soc. 1990, 112, 3209–3210. doi:10.1021/ja00164a051 |
| 32. | Miller, D. J.; Gao, J.; Truhlar, D. G.; Young, N. J.; Gonzalez, V.; Allemann, R. K. Org. Biomol. Chem. 2008, 6, 2346–2354. doi:10.1039/b804198a |
| 33. | Surendra, K.; Corey, E. J. J. Am. Chem. Soc. 2008, 130, 8865–8869. doi:10.1021/ja802730a |
| 73. | Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675. doi:10.1063/1.475428 |
| 74. | Matsuda, S. P. T.; Wilson, W. K.; Xiong, Q. Org. Biomol. Chem. 2006, 4, 530–543. doi:10.1039/b513599k |
| 17. | We computed the geometry of hydrogenated 1, and the carbon skeleton did not relax much upon hydrogenation (see Supporting Information File 1). |
| 8. |
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements. |
| 66. | Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1990, 94, 5523–5527. doi:10.1021/j100377a021 |
| 67. | Fukui, K. Acc. Chem. Res. 1981, 14, 363–368. doi:10.1021/ar00072a001 |
| 11. |
Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v
And references therein. |
| 12. | Jain, R.; Bally, T.; Rablen, P. R. J. Org. Chem. 2009, 74, 4017–4023. doi:10.1021/jo900482q |
| 13. | CHESHIRE, Chemical Shift Repository with Coupling Constants Added Too. http://cheshirenmr.info. |
| 14. | Lodewyk, M. W.; Tantillo, D. J. J. Nat. Prod. 2011, 74, 1339–1343. doi:10.1021/np2000446 |
| 15. | Quasdorf, K. W.; Huters, A. D.; Lodewyk, M. W.; Tantillo, D. J.; Garg, N. K. J. Am. Chem. Soc. 2012, 134, 1396–1399. doi:10.1021/ja210837b |
| 16. | Lodewyk, M. W.; Soldi, C.; Jones, P. B.; Olmstead, M. M.; Rita, J.; Shaw, J. T.; Tantillo, D. J. J. Am. Chem. Soc. 2012, 134, 18550–18553. doi:10.1021/ja3089394 |
| 1. | Zu, L.; Xu, M.; Lodewyk, M. W.; Cane, D. E.; Peters, R. J.; Tantillo, D. J. J. Am. Chem. Soc. 2012, 134, 11369–11371. doi:10.1021/ja3043245 |
| 8. |
Tantillo, D. J. Nat. Prod. Rep. 2011, 28, 1035–1053. doi:10.1039/c1np00006c
See for leading references on quantum chemical calculations on terpene-forming carbocation rearrangements. |
| 18. | Gutta, P.; Tantillo, D. J. J. Am. Chem. Soc. 2006, 128, 6172–6179. doi:10.1021/ja058031n |
| 19. | Wang, S. C.; Tantillo, D. J. Org. Lett. 2008, 10, 4827–4830. doi:10.1021/ol801898v |
| 20. | Gutta, P.; Tantillo, D. J. Angew. Chem., Int. Ed. 2005, 44, 2719–2723. doi:10.1002/anie.200461915 |
| 21. | Tantillo, D. J. J. Phys. Org. Chem. 2008, 21, 561–570. doi:10.1002/poc.1320 |
| 22. | Tantillo, D. J. Chem. Soc. Rev. 2010, 39, 2847–2854. doi:10.1039/b917107j |
| 68. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 69. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 70. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 71. | Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623–11627. doi:10.1021/j100096a001 |
| 72. | Tirado-Rives, J.; Jorgensen, W. L. J. Chem. Theory Comput. 2008, 4, 297–306. doi:10.1021/ct700248k |
| 9. |
Suyama, T. L.; Gerwick, W. H.; McPhail, K. L. Bioorg. Med. Chem. 2011, 19, 6675–6701. doi:10.1016/j.bmc.2011.06.011
Leading references on misassigned natural product structures. |
| 10. |
Nicolaou, K. C.; Snyder, S. A. Angew. Chem., Int. Ed. 2005, 44, 1012–1044. doi:10.1002/anie.200460864
Leading references on misassigned natural product structures. |
| 55. | Hong, Y. J.; Tantillo, D. J. Org. Biomol. Chem. 2010, 8, 4589–4600. doi:10.1039/c0ob00167h |
| 58. |
Roy, A.; Roberts, F. G.; Wilderman, P. R.; Zhou, K.; Peters, R. J.; Coates, R. M. J. Am. Chem. Soc. 2007, 129, 12453–12460. doi:10.1021/ja072447e
Discussions of pyrophosphate as a possible active site base in terpene synthases. |
| 59. | Greenhagen, B. T.; O’Maille, P. E.; Noel, J. P.; Chappell, J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9826–9831. doi:10.1073/pnas.0601605103 |
| 60. | Shishova, E. Y.; Di Costanzo, L.; Cane, D. E.; Christianson, D. W. Biochemistry 2007, 46, 1941–1951. doi:10.1021/bi0622524 |
| 61. | Peters, R. J.; Croteau, R. B. Arch. Biochem. Biophys. 2003, 417, 203–211. doi:10.1016/S0003-9861(03)00347-3 |
| 62. | Garms, S.; Chen, F.; Boland, W.; Gershenzon, J.; Köllner, T. G. Phytochemistry 2012, 75, 6–13. doi:10.1016/j.phytochem.2011.12.009 |
| 63. | Zhou, K.; Peters, R. J. Chem. Commun. 2011, 47, 4074–4080. doi:10.1039/c0cc02960b |
| 64. | Miller, D. J.; Allemann, R. K. Nat. Prod. Rep. 2012, 29, 60–71. doi:10.1039/c1np00060h |
| 2. | Barrow, C. J.; Blunt, J. W.; Munro, M. H. G. Aust. J. Chem. 1988, 41, 1755–1761. doi:10.1071/CH9881755 |
| 2. | Barrow, C. J.; Blunt, J. W.; Munro, M. H. G. Aust. J. Chem. 1988, 41, 1755–1761. doi:10.1071/CH9881755 |
| 21. | Tantillo, D. J. J. Phys. Org. Chem. 2008, 21, 561–570. doi:10.1002/poc.1320 |
| 37. | Williams, A. Concerted Organic and Bio-Organic Mechanisms; CRC Press: Boca Raton, FL, USA, 2000. |
| 38. | Dewar, M. J. S. J. Am. Chem. Soc. 1984, 106, 209–219. doi:10.1021/ja00313a042 |
| 11. |
Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v
And references therein. |
| 12. | Jain, R.; Bally, T.; Rablen, P. R. J. Org. Chem. 2009, 74, 4017–4023. doi:10.1021/jo900482q |
| 13. | CHESHIRE, Chemical Shift Repository with Coupling Constants Added Too. http://cheshirenmr.info. |
| 73. | Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664–675. doi:10.1063/1.475428 |
| 74. | Matsuda, S. P. T.; Wilson, W. K.; Xiong, Q. Org. Biomol. Chem. 2006, 4, 530–543. doi:10.1039/b513599k |
| 35. | Hoffmann, R.; Woodward, R. B. Acc. Chem. Res. 1968, 1, 17–22. doi:10.1021/ar50001a003 |
| 36. | Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811 |
| 11. |
Lodewyk, M. W.; Siebert, M. R.; Tantillo, D. J. Chem. Rev. 2012, 112, 1839–1862. doi:10.1021/cr200106v
And references therein. |
| 12. | Jain, R.; Bally, T.; Rablen, P. R. J. Org. Chem. 2009, 74, 4017–4023. doi:10.1021/jo900482q |
| 13. | CHESHIRE, Chemical Shift Repository with Coupling Constants Added Too. http://cheshirenmr.info. |
| 20. | Gutta, P.; Tantillo, D. J. Angew. Chem., Int. Ed. 2005, 44, 2719–2723. doi:10.1002/anie.200461915 |
| 49. | McMurry, J. E.; Lectka, T. Acc. Chem. Res. 1992, 25, 47–53. doi:10.1021/ar00013a007 |
| 50. | Ponec, R.; Yuzhakov, G.; Tantillo, D. J. J. Org. Chem. 2004, 69, 2992–2996. doi:10.1021/jo035506p |
| 51. | Tantillo, D. J.; Hoffmann, R. J. Am. Chem. Soc. 2003, 125, 4042–4043. doi:10.1021/ja021394s |
| 47. | D-type structures arising from alternative conformers of C were examined, but these correspond to nonproductive stereoisomers; see Supporting Information File 1 for details. |
| 20. | Gutta, P.; Tantillo, D. J. Angew. Chem., Int. Ed. 2005, 44, 2719–2723. doi:10.1002/anie.200461915 |
| 22. | Tantillo, D. J. Chem. Soc. Rev. 2010, 39, 2847–2854. doi:10.1039/b917107j |
| 48. | Ponec, R.; Bultinck, P.; Gutta, P.; Tantillo, D. J. J. Phys. Chem. A 2006, 110, 3785–3789. doi:10.1021/jp0568430 |
| 41. | Geometries in which the two methyl groups pointed in opposite directions were also examined, but the resulting structures were not productive for caryolene formation; see Supporting Information File 1 for details. |
| 42. | Grob, C. A. Acc. Chem. Res. 1983, 16, 426–431. doi:10.1021/ar00096a001 |
| 43. | Brown, H. C. Acc. Chem. Res. 1983, 16, 432–440. doi:10.1021/ar00096a002 |
| 44. | Olah, G. A.; Prakash, G. K. S.; Saunders, M. Acc. Chem. Res. 1983, 16, 440–448. doi:10.1021/ar00096a003 |
| 45. | Walling, C. Acc. Chem. Res. 1983, 16, 448–454. doi:10.1021/ar00096a004 |
| 46. |
Brown, H. C. The Nonclassical Ion Problem; Plenum Press: New York, NY, 1977.
With comments by P. v. R. Schleyer. |
| 39. | Nouri, D. H.; Tantillo, D. J. Tetrahedron 2008, 64, 5672–5679. doi:10.1016/j.tet.2008.04.028 |
| 40. | Sio, V.; Harrison, J. G.; Tantillo, D. J. Tetrahedron 2012, 53, 6919–6922. doi:10.1016/j.tetlet.2012.10.019 |
© 2013 Nguyen and Tantillo; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)