Abstract
γ-Hydroxybutanoic acid (GHB) is used as a date-rape drug, which renders the victims unconscious and defenceless. Intoxications are very difficult to detect for forensic scientists due to rapid metabolism to endogenous levels of GHB. We recently discovered a new major metabolite, 2, of GHB (1) that could potentially extend the analytical detection window for GHB intoxications. Herein we disclose synthetic procedures based on a Koenigs–Knorr glucuronidation approach that provides GHB glucuronide 2 and a deuterium-labelled analogue d4-2 of high purity suitable for analytical chemistry. In addition, we have assessed the stability of GHB glucuronide 2 by mimicking the natural pH range for urine, which is of importance in the development of new analytical methods. Using NMR we show that GHB glucuronide 2 is highly stable towards aqueous hydrolysis within the pH range normally observed for urine even at elevated temperature.

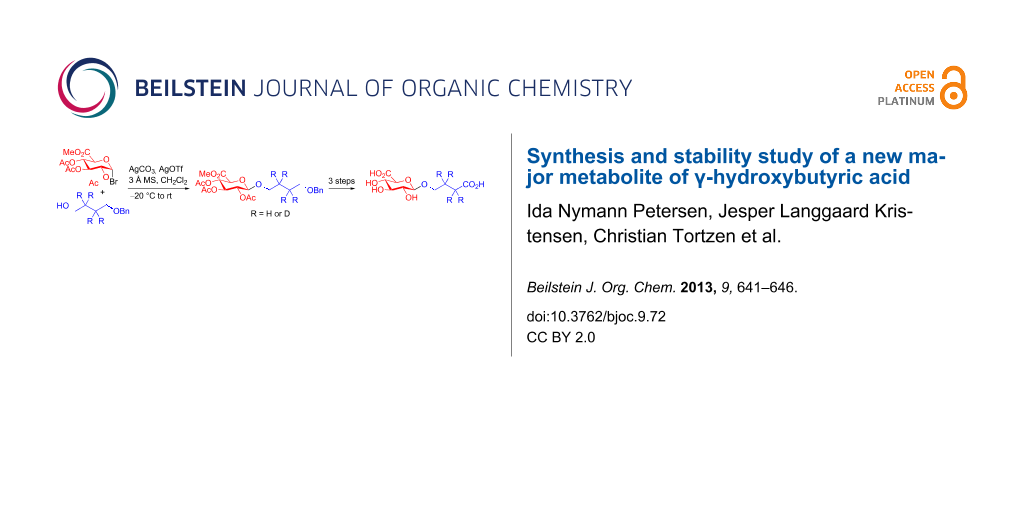
Graphical Abstract
Introduction
The abuse of illicit drugs continues to be a very significant problem to society and results in many drug-related accidents and deaths worldwide. Law enforcement agencies require the assistance of analytical laboratories to identify drugs from a wide variety of sources in order to try and combat this problem. Despite huge advances in analytical sciences certain illegal drugs continue to elude analytical detection. γ-Hydroxybutanoic acid (GHB, 1, Figure 1), often referred to as Fantasy or liquid ecstasy, is a so-called predatory drug or date-rape drug.
![[1860-5397-9-72-1]](/bjoc/content/figures/1860-5397-9-72-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Hypothesised glucuronidation of GHB (1) by UDP-glucuronosyltransferase to give glucuronide 2. UDP = Uridinediphosphate.
Figure 1: Hypothesised glucuronidation of GHB (1) by UDP-glucuronosyltransferase to give glucuronide 2. UDP =...
Most commonly, the ingestion of GHB renders the victim unconscious and defenceless due to the heavy sedative effect, and GHB often induces short-term memory loss in victims thereby complicating case prosecution. GHB is also frequently used as a recreational drug [1] with a high risk of fatal overdosing and with a high incidence of toxic effects, including impaired consciousness, coma and numerous reports on acute poisonings and drug-related deaths [2]. After consumption, GHB is rapidly metabolised in vivo and is only detectable above endogenous levels in a narrow time window of 3–6 h [3,4]. Less than 1% of GHB is excreted unchanged in urine, and current analytical methods for serum or urine continue to be problematic. Confirmed positive laboratory samples for GHB intoxications are relatively rare, either due to delayed sampling or simply because samples are not forwarded to a toxicology laboratory [2]. Consequently any analytical method that could extend the analytical detection window for GHB would represent a very important advance in analytical and forensic science with immediate implications for society.
UDP-glucuronosyltransferase is an important enzyme in the metabolism of xenobiotics that transforms functional groups such as alcohols and carboxylic acids to their respective glucuronides (e.g., Figure 1). Glucuronides generally have longer plasma half-life values than the unmodified compound (e.g., ethyl glucuronide versus ethanol), making it possible to use the glucuronide as a biomarker to extend the analytical detection window [5]. By analogy with ethanol, we hypothesised the existence of a GHB glucuronide, and recently discovered that GHB glucuronide 2 is indeed a major metabolite of GHB (Figure 1) [6]. The presence of GHB glucuronide 2 is likely to have important implications for future analysis of GHB in clinical and forensic toxicology. The mono-sodium salt of GHB glucuronide 2 made by chemical synthesis is commercially available from Reseachem (http://www.reseachem.ch), but an isotope-labelled analogue is not available. To the best of our knowledge the synthesis or use of compound 2 has never been reported.
Herein we wish to disclose the synthesis of GHB glucuronide 2 and a deuterium labelled analogue d4-2, which is required as an internal standard for chromatography. Moreover, we have assessed the stability of GHB glucuronide 2 towards aqueous hydrolysis within the pH range normally observed for urine, which is of importance in the development of new analytical methods.
Results and Discussion
Synthesis and stability assessment
Synthesis of GHB glucuronides 2 and d4-2
The synthesis of small molecule glucuronide derivatives can be carried out by a wide variety of synthetic [7,8] and biocatalytic [9,10] methods. Initially, we favoured a synthetic approach using Schmidt trichloroacetimidate chemistry [11] with trichloroacetimidate donor 3 (Scheme 1) that has been used successfully by others for the synthesis of alcohol glucuronides [7,8,12]. Moreover, the required trichloroacetimidate donor 3 is stable and accessible from commercially available glucuronolactone by using literature methods (Scheme 1) [13-16]. We anticipated that glucuronidation with a mono-protected 1,4-butanediol acceptor [17-19] would be feasible and that it would be possible to deprotect and oxidise the glucuronidation product (4 or 5) to provide target molecule 2.
![[1860-5397-9-72-i1]](/bjoc/content/inline/1860-5397-9-72-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schmidt glucuronidation [11] with trichloroacetimidate 3. Synthesis of 4 and 5 using acceptors 7 and 8 was attempted several times by using BF3·OEt2, 3 Å MS, CH2Cl2, −20 °C to rt, and TMSOTf, 3 Å MS, CH2Cl2, −20 °C to rt, but never gave any of the desired material. aConversion to 6 with acceptor 9 was judged to be >80% by 1H NMR analysis of the crude product after work-up. TBDPS: tert-butyldiphenylsilyl; MS: molecular sieves.
Scheme 1: Schmidt glucuronidation [11] with trichloroacetimidate 3. Synthesis of 4 and 5 using acceptors 7 and 8 ...
However, attempts under commonly employed reaction conditions for glucuronidation returned none of the desired product 4 or 5. Glucuronidation of alcohols with trichloroacetimidate 3 has been reported to be problematic due to the high reactivity of the acceptor relative to the donor resulting in trans-esterification [20-23]. Indeed in our case acetylated acceptor was the only identified product from the reaction. To evaluate whether the high reactivity of the acceptor was the problem we tested the less reactive acceptor 4-benzyloxybutanoic acid (9). As anticipated a less reactive acceptor provided the glucuronidated product 6 in high yield as estimated by 1H NMR on the crude reaction mixture. Trans-esterification during glucuronidation can be suppressed by changing from acetyl protection on the sugar moiety to less reactive benzoyl, isobutyroyl or pivaloyl protection groups [21-23]. Alternatively, the use of bromo-derivative 10 (Scheme 2), which is easily synthesised in two steps from glucuronolactone [14,24] has been shown to glucuronidate primary and secondary alcohols under Koenigs–Knorr conditions [7,8,25,26].
![[1860-5397-9-72-i2]](/bjoc/content/inline/1860-5397-9-72-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Koenigs–Knorr glucuronidation [27] with bromide 10 and acceptors 7 and 8.
Scheme 2: Koenigs–Knorr glucuronidation [27] with bromide 10 and acceptors 7 and 8.
Due to the easy access of donor 10 from glucuronolactone we decided to explore the Koenigs–Knorr glucuronidation route [27]. Using standard Koenigs–Knorr conditions donor 10 does indeed glucuronidate acceptor 8 to give the desired product 5 albeit only in 30% yield. Unfortunately, removal of the TBDPS protection group to provide the desired alcohol 11 proved difficult and complex mixtures were obtained on using both TBAF in THF and HF in pyridine. Fortunately, glucuronidation also proceeded with acceptor 7 to give 4, and in this case the benzyl group was easily removed by catalytic hydrogenation to provide alcohol 11 in good yield. Oxidation of alcohol 11 was carried out similarly to that reported elsewhere [19], using Epp and Widlanski’s TEMPO oxidation procedure [28] to furnish carboxylic acids 12 and d4-12 (Scheme 3). Finally, deprotection under basic condition followed by treatment with an acidic ion-exchange resin provided the required GHB glucuronides 2 and d4-2 in good yield.
![[1860-5397-9-72-i3]](/bjoc/content/inline/1860-5397-9-72-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of GHB glucuronides 2 and d4-2 by using a Koenigs–Knorr glucuronidation approach. TEMPO: 2,2,6,6-tetramethyl-1-piperidinyloxyl, BAIB: [bis(acetoxy)iodo]benzene.
Scheme 3: Synthesis of GHB glucuronides 2 and d4-2 by using a Koenigs–Knorr glucuronidation approach. TEMPO: ...
1H NMR analysis of d4-2 showed the complete absence of methylene groups b and c (Figure 2). In addition, analysis of d4-2 by mass spectrometry showed the presence of less than 0.14% of 2, thus satisfying the demand for a highly pure internal standard [6].
![[1860-5397-9-72-2]](/bjoc/content/figures/1860-5397-9-72-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectrum (D2O, 300 MHz) of GHB glucuronides 2 (top) and d4-2 (bottom). As anticipated, methylene protons b and c are absent in d4-2 (cf. labelling in Scheme 3).
Figure 2: 1H NMR spectrum (D2O, 300 MHz) of GHB glucuronides 2 (top) and d4-2 (bottom). As anticipated, methy...
Stability assessment of GHB glucuronide 2 by NMR
The stability of GHB glucuronide 2 is critical if it is to be used for routine analysis by analytical and forensic chemists. Consequently, a series of NMR experiments to assess the stability of GHB glucuronide 2 were conducted. To mimic the normal pH range for urine (pH 4.6–8) mono- and a di-basic sodium phosphate buffers were employed as NMR solvents to give pH values of 4.8 and 9.0, respectively (Supporting Information File 1). The stability of GHB glucuronide 2 was assessed from 18 to 90 °C for several days. GHB glucuronide 2 was found to be almost completely stable in both buffer systems over the entire temperature range. Only after heating at 90 °C in acidic buffer for 3 days could a small amount of γ-butyrolactone (GBL) be detected (Figure 3). Under forcing acidic conditions (autoclaving for 15 min with 4 M aq HCl) GHB glucuronide 2 was completely degraded whilst being stable towards strong base (3 M aq NaOH) [6].
![[1860-5397-9-72-3]](/bjoc/content/figures/1860-5397-9-72-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra (500 MHz) of GHB glucuronide 2 in pH 4.8 buffer at t = 0 (rt) and t = 72 h (90 °C) by using a Watergate-type water suppression method (Supporting Information File 1). After heating at 90 °C for 72 h GBL starts to form at low concentration (indicated with arrows).
Figure 3: 1H NMR spectra (500 MHz) of GHB glucuronide 2 in pH 4.8 buffer at t = 0 (rt) and t = 72 h (90 °C) b...
Conclusion
Herein we have described the synthesis of a recently discovered major metabolite of GHB that has the potential to extend the analytical detection window for GHB intoxications significantly. GHB glucuronide 2 and the isotope-labelled analogue d4-2 were shown to be of sufficient purity for use in analytical laboratories. Moreover, the stability of GHB glucuronide 2 was assessed under basic and acidic conditions mimicking the pH range typically observed in urine samples. GHB glucuronide was demonstrated to be highly stabile towards aqueous hydrolysis within the pH range normally observed for urine even at elevated temperature for several days, making it suitable for method development within analytical and forensic chemistry.
Experimental
General
For reactions conducted under anhydrous conditions, glassware was dried overnight in an oven at 150 °C and was allowed to cool in a desiccator over anhydrous KOH. Anhydrous reactions were carried out under nitrogen. THF was distilled from sodium wire with benzophenone as indicator. Dichloromethane and pyridine were dried and stored over 4 Å molecular sieves. Thin-layer chromatography (TLC) was carried out on commercially available precoated aluminium sheets (Merck 60 F254). The quoted Rf values are rounded to the nearest 0.05. 1H and 13C NMR was run on a Varian Mercury 300 MHz, a Varian Gemini 300 MHz and a Bruker 500 MHz Avance III Fourier transform NMR spectrometer, respectively, by using an internal deuterium lock. Solvents were used as internal standard when assigning NMR spectra [29]. J values are given in hertz (Hz) and rounded to the nearest 0.5 Hz. Dry column vacuum chromatography (DCVC) was carried out according to the published procedure [30]. High-resolution mass spectra were recorded on a Micromass Q-TOF 1.5, UB137. Melting points were recorded on an OptiMelt MPA100 from Stanford Research Systems.
Glucuronide donors 10 [24] and 3 [13-16] and acceptors 8, 7 and d4-7 [17-19] were synthesised according to literature procedures. All analytical data were in agreement with those previously published.
Methyl 2,3,4-tri-O-acetyl-1-O-(1-hydroxybut-4-yl)-β-D-glucopyranosiduronate (11): Bromide 10 (0.6 g, 1.17 mmol) and 4-benzyloxybutan-1-ol (7) (0.3 g, 1.17 mmol) were dissolved in anhydrous CH2Cl2 (10 mL) and stirred with molecular sieves (3 Å) for 1 h. The reaction mixture was cooled to −20 °C before AgOTf (0.43 g, 1.17 mmol) and Ag2CO3 (0.46 g, 1.17 mmol) were added. The mixture was stirred for 3 h at −20 °C and the solids were removed by filtration through a pad of Celite. Sat. aq NaHCO3 solution (50 mL) was added, and the mixture was extracted with EtOAc (3 × 40 mL). The combined organic phases were dried (MgSO4), filtered and evaporated in vacuo. The residue was purified by DCVC [id 2 cm; 20 mL fractions 20% EtOAc in n-heptane (100 mL) (v/v); 50% EtOAc in n-heptane (100 mL) (v/v)] to give glucuronide 4 (0.48 g) contaminated with bromide 10 and alcohol 7. With no further purification the mixture was dissolved in MeOH (19 mL) and Pd on activated charcoal (10% w/w, 20 mg) was added, and then the flask was fitted with a H2 balloon and stirred vigorously. After 24 h the mixture was filtered through a pad of Celite and concentrated in vacuo. The residue was purified by DCVC [id 4 cm; 20 mL fractions 20% EtOAc in n-heptane (100 mL) (v/v); 80% EtOAc in n-heptane (100 mL) (v/v)] to give glucuronide 11 (0.16 g, 50%) as colourless plates. Mp: 78.4 °C (from EtOAc, n-heptane); ![[Graphic 1]](/bjoc/content/inline/1860-5397-9-72-i4.svg?max-width=637&scale=1.18182) −80.0 (c 0.5, MeOH); Rf 0.3 (80% EtOAc in n-heptane, v/v); IR (CHCl3) νmax: 3300 (OH), 1724 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C17H26O11Na, 429.1373; found, 429.1392; 1H NMR (300 MHz, CDCl3) δ 5.29–5.17 (m, 2H, H3 and H4), 5.06–4.46 (m, 1H, H2), 4.50 (d, J = 8 Hz, 1H, H1), 4.05 (m, 1H, H4), 3.97 (m, 1H, H6), 3.76 (s, 3H, Me), 3.66 (q, J = 6 Hz, 2H, H9), 3.56 (m, 1H, H6), 2.07 (s, 3H, Ac), 2.04 (2 s, 6H, 2 × Ac), 1.65 (m, 4H, H7 and H8); 13C NMR (300 MHz, CDCl3) δ 170.1, 169.4, 169.3, 167.2 (4 × C=O), 100.8 (C1), 72.7, 72.1, 71.4, 70.3, 69.5 (4 × CH and 1 × CH2), 62.5 (CH2OH), 53.1 (CO2CH3), 29.4, 26.0 (2 × CH2), 20.9 (2 × Ac), 20.8 (Ac).
−80.0 (c 0.5, MeOH); Rf 0.3 (80% EtOAc in n-heptane, v/v); IR (CHCl3) νmax: 3300 (OH), 1724 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C17H26O11Na, 429.1373; found, 429.1392; 1H NMR (300 MHz, CDCl3) δ 5.29–5.17 (m, 2H, H3 and H4), 5.06–4.46 (m, 1H, H2), 4.50 (d, J = 8 Hz, 1H, H1), 4.05 (m, 1H, H4), 3.97 (m, 1H, H6), 3.76 (s, 3H, Me), 3.66 (q, J = 6 Hz, 2H, H9), 3.56 (m, 1H, H6), 2.07 (s, 3H, Ac), 2.04 (2 s, 6H, 2 × Ac), 1.65 (m, 4H, H7 and H8); 13C NMR (300 MHz, CDCl3) δ 170.1, 169.4, 169.3, 167.2 (4 × C=O), 100.8 (C1), 72.7, 72.1, 71.4, 70.3, 69.5 (4 × CH and 1 × CH2), 62.5 (CH2OH), 53.1 (CO2CH3), 29.4, 26.0 (2 × CH2), 20.9 (2 × Ac), 20.8 (Ac).
Methyl 2,3,4-tri-O-acetyl-1-O-(1-carboxyprop-3-yl)-β-D-glucopyranosiduronate (12): Alcohol 11 (0.16 g, 0.4 mmol), 2,2,6,6-tetramethylpiperinyloxyl (12.5 mg, 0.08 mmol) and [bis(acetoxy)iodo]benzene (0.28 g, 0.88 mmol) were dissolved in H2O/CH3CN (1 mL). After 12 h water (20 mL) was added and the mixture was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with water (40 mL), dried (MgSO4) and evaporated in vacuo. The residue was purified by DCVC [id 2 cm; 20 mL fractions 25% EtOAc in n-heptane (100 mL); 75% EtOAc in n-heptane (100 mL) (v/v)] to give carboxylic acid 12 (0.10 g, 62%) as colourless needles. −22.0 (c 1, MeOH); IR (CHCl3) νmax: 3399 (O-H) and 1754 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C17H24O12Na, 443.1165; found, 443.1181; 1H NMR (300 MHz, CDCl3) δ 5.29–5.17 (m, 2H, H3 and H4), 5.06–4.46 (m, 1H, H2), 4.5 (d, J = 8 Hz, 1H, H1), 4.05 (m, 1H, H5), 3.97 (m, 1H, H6), 3.76 (s, 3H, Me), 3.56 (m, 1H, H6), 2.45 (t, J = 7 Hz, 2H, H8), 2.07 (s, 3H, Ac), 2.04 (2 s, 6H, 2 × Ac), 1.93 (m, 2H, H7); 13C NMR (300 MHz, CDCl3) δ 178.7 (CO2H), 170.1, 169.4, 169.3, 167.2 (4 × C=O), 100.8 (C1), 72.7, 72.1, 71.3, 69.5, 69.0 (4 × CH and 1 × CH2), 53.1 (CO2CH3), 30.3 (CH2), 24.6 (CH2), 20.8 (2 × Ac), 20.7 (Ac).
1-O-(3-Carboxypropyl)-β-D-glucopyranosiduronic acid (2): Carboxylic ester 12 (0.14 g, 0.33 mmol) was dissolved in water (4 mL) and methanol (12 mL) before Na2CO3 (0.21 g, 2 mmol) was added. After 2 days water (2 mL) and glacial acetic acid (0.1 mL) were added. The mixture was filtered through a short column of Dowex-50 resin (prewashed with 3 mL MeOH), and the resin was washed with water (10 mL). The solvents were evaporated in vacuo to give carboxylic acid 2 (72 mg, 77%) as a clear gum that required no further purification. −44.0 (c 1, H2O); Rf 0.45 (1:1:1:1 EtOAc/n-butanol/acetic acid/water, v/v/v/v); IR (CHCl3) νmax: 3400 (O-H) and 1715 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C10H16O9Na, 303.0692; found, 303.0694; 1H NMR (300 MHz, CDCl3) δ 4.31 (d, J = 7.5 Hz, 1H, H1), 3.8 (m, 1H, H5), 3.7 (dt, J = 10.0, 6.5 Hz, 1H, H6), 3.5 (dt, J = 10.0, 6.5 Hz, 1H, H6), 3.35 (m, 2H, H3 and H4), 3.1 (m, 1H, H2), 2.3 (t, J = 7.4 Hz, 2H, H8), 1.72 (m, 2H, H7); 13C NMR (300 MHz, CDCl3) δ 178.4, 172.3 (2 × C=O), 102.5 (C1), 75.5, 74.7, 73.0, 71.6, 69.8 (4 × CH and 1 × CH2), 30.6 (CH2C=O), 24.7 (CH2CH2C=O).
Methyl 2,3,4-tri-O-acetyl-1-O-(2,3-[2H4]-1-hydroxybut-4-yl)-β-D-glucopyransiduronate (d4-11): Prepared as described above to give alcohol d4-11 (407 mg, 50%) as white needles. +23.0 (c 1, MeOH); Rf 0.25 (1:1 EtOAc/n-heptane, v/v); IR (CHCl3) νmax: 3399 (O-H) and 1754 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C17H22D4O11Na, 433.1625; found, 433.1634; 1H NMR (300 MHz, CDCl3) δ 5.29–5.17 (m, 2H, H3 and H4), 5.06–4.46 (m, 1H, H2), 4.55 (d, J = 8 Hz, 1H, H1), 4.05 (m, 1H, H4), 3.94 (d, J = 10 Hz, 1H, H6), 3.76 (s, 3H, Me), 3.66 (s, 2H, H9), 3.56 (d, J = 10 Hz, 1H, H6), 2.07 (s, 3H, Ac), 2.04 (2 s, 6H, 2 × Ac); 13C NMR (300 MHz, CDCl3) δ 170.5, 169.7, 169.6, 167.6 (4 × C=O), 101.2 (C1), 73.0, 72.5, 71.7, 70.5, 69.9 (4 × CH and 1 × CH2), 62.7 (CH2OH), 53.4 (CO2CH3), 43.8 (m, CD2CH2OH), 25.5 (m, CD2), 21.2 (2 × Ac), 21.1 (Ac).
Methyl 2,3,4-tri-O-acetyl-(1,2-[2H4]-1-carboxyprop-3-yl)-β-D-glucopyransiduronate (d4-12): Prepared as described above to give carboxylic acid d4-12 (140 mg, 43%) as white needles. +21.8 (c 1, MeOH); IR (CHCl3) νmax: 3399 (O-H) and 1754 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C17H22D4O11Na, 447.1418; found, 447.1374; 1H NMR (300 MHz, CDCl3) δ 5.29–5.17 (m, 2H, H3 and H4), 5.06–4.46 (m, 1H, H2), 4.55 (d, J = 8 Hz, 1H, H1), 4.05 (m, 1H, H4), 3.94 (d, J = 10 Hz, 1H, H6), 3.76 (s, 3H, Me), 3.66 (s, 2H, H9), 3.56 (d, J = 10 Hz, 1H, H6), 2.07 (s, 3H, Ac), 2.04 (2 s, 6H, 2 × Ac); 13C NMR (300 MHz, CDCl3) δ 178.8 (CO2H), 170.5, 169.7, 169.7, 167.5 (4 × C=O), 101.1 (C1), 73.0, 72.5, 71.6, 69.9, 69.1 (4 × CH and 1 × CH2), 53.4 (CO2CH3), 29.8 (m, CD2C=O), 24.2 (m, CD2CD2C=O), 20.8 (2 × Ac), 20.7 (Ac).
1-O-(1,2-[2H4]-1-Carboxyprop-3-yl)-β-D-glucopyranosiduronic acid (d4-2): Prepared as described above to give carboxylic acid d4-2 (55 mg, 77%) as a clear colourless gum. −46.0 (c 1, H2O); Rf 0.45 (1:1:1:1 EtOAc/n-butanol/acetic acid/water, v/v/v/v); IR (CHCl3) νmax: 3400 (O-H) and 1715 (C=O) cm−1; MS (ESI+) m/z: [M + Na]+ calcd for C10H12D4O9Na, 307.095; found, 307.0951; 1H NMR (300 MHz, CDCl3) δ 4.32 (d, J = 8 Hz, 1H, H1), 3.8 (m, 1H, H5), 3.7 (d, J = 10 Hz, 1H, H6), 3.5 (d, J = 10, 6.5 Hz, 1H, H6), 3.37 (m, 2H, H3 and H4), 3.1 (m, 1H, H2); 13C NMR (300 MHz, CDCl3) δ 178.3, 172.3 (2 × C=O), 102.5 (C1), 75.5, 74.7, 73.0, 71.6, 69.8 (4 × CH and 1 × CH2), 30.0 (m, CD2C=O), 23.8 (m, CD2CD2C=O).
Supporting Information
| Supporting Information File 1: 1D and 2D NMR spectra for 2 and d4-2 and all details for the NMR stability study of GHB glucuronide 2. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
Carter, L. P.; Pardi, D.; Gorsline, J.; Griffiths, R. R. Drug Alcohol Depend. 2009, 104, 1–10. doi:10.1016/j.drugalcdep.2009.04.012
Return to citation in text: [1] -
Knudsen, K.; Greter, J.; Verdicchio, M. Clin. Toxicol. 2008, 46, 187–192. doi:10.1080/15563650701263633
Return to citation in text: [1] [2] -
Haller, C.; Thai, D.; Jacob, P. I.; Dyer, J. E. J. Anal. Toxicol. 2006, 30, 360–364.
Return to citation in text: [1] -
Brailsford, A. D.; Cowan, D. A.; Kicman, A. T. J. Anal. Toxicol. 2012, 36, 88–95. doi:10.1093/jat/bkr023
Return to citation in text: [1] -
Jatlow, P.; O'Malley, S. S. Alcohol.: Clin. Exp. Res. 2010, 34, 968–975. doi:10.1111/j.1530-0277.2010.01171.x
Return to citation in text: [1] -
Petersen, I. N.; Kristensen, J. L.; Tortzen, C.; Pedersen, D. S.; Breindahl, T. J. Anal. Toxicol. 2013, in press.
Return to citation in text: [1] [2] [3] -
Stachulski, A. V.; Jenkins, G. N. Nat. Prod. Rep. 1998, 15, 173–186. doi:10.1039/a815173y
Return to citation in text: [1] [2] [3] -
Kaspersen, F. M.; van Boeckel, C. A. A. Xenobiotica 1987, 17, 1451–1471. doi:10.3109/00498258709044005
Return to citation in text: [1] [2] [3] -
Wilkinson, S. M.; Liew, C. W.; Mackay, J. P.; Salleh, H. M.; Withers, S. G.; McLeod, M. D. Org. Lett. 2008, 10, 1585–1588. doi:10.1021/ol8002767
Return to citation in text: [1] -
Khymenets, O.; Joglar, J.; Clapés, P.; Parella, T.; Covas, M.-I.; de la Torre, R. Adv. Synth. Catal. 2006, 348, 2155–2162. doi:10.1002/adsc.200606221
Return to citation in text: [1] -
Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311
Return to citation in text: [1] [2] -
Pews-Davtyan, A.; Pirojan, A.; Shaljyan, I.; Awetissjan, A. A.; Reinke, H.; Vogel, C. J. Carbohydr. Chem. 2003, 22, 939–962. doi:10.1081/CAR-120026603
Return to citation in text: [1] -
Brown, R. T.; Scheinmann, F.; Stachulski, A. V. J. Chem. Res., Synop. 1997, 370–371.
Return to citation in text: [1] [2] -
Bollenback, G. N.; Long, J. W.; Benjamin, D. G.; Lindquist, J. A. J. Am. Chem. Soc. 1955, 77, 3310–3315. doi:10.1021/ja01617a047
Return to citation in text: [1] [2] [3] -
Trynda, A.; Madaj, J.; Konitz, A.; Wiśniewski, A. Carbohydr. Res. 2000, 329, 249–252. doi:10.1016/S0008-6215(00)00157-9
Return to citation in text: [1] [2] -
Dumont-Hornebeck, B. A.; Joly, J.-P.; Coulon, J.; Chapleur, Y. Carbohydr. Res. 1999, 320, 147–160. doi:10.1016/S0008-6215(99)00151-2
Return to citation in text: [1] [2] -
Djerassi, C.; Sheehan, M.; Spangler, R. J. J. Org. Chem. 1971, 36, 3526–3532. doi:10.1021/jo00822a013
Return to citation in text: [1] [2] -
George, S.; Sudalai, A. Tetrahedron: Asymmetry 2007, 18, 975–981. doi:10.1016/j.tetasy.2007.04.008
Return to citation in text: [1] [2] -
Raunkjær, M.; Pedersen, D. S.; Elsey, G. M.; Sefton, M. A.; Skouroumounis, G. K. Tetrahedron Lett. 2001, 42, 8717–8719. doi:10.1016/S0040-4039(01)01890-1
Return to citation in text: [1] [2] [3] -
Berrang, B.; Brine, G. A.; Carroll, F. I. Synthesis 1997, 1165–1168. doi:10.1055/s-1997-3187
Return to citation in text: [1] -
Brown, R. T.; Carter, N. K.; Lumbard, K. W.; Scheinmann, F. Tetrahedron Lett. 1995, 36, 8661–8664. doi:10.1016/0040-4039(95)01786-H
Return to citation in text: [1] [2] -
Brown, R. T.; Carter, N. E.; Mayalarp, S. P.; Scheinmann, F. Tetrahedron 2000, 56, 7591–7594. doi:10.1016/S0040-4020(00)00664-5
Return to citation in text: [1] [2] -
Lucas, R.; Alcantara, D.; Morales, J. C. Carbohydr. Res. 2009, 344, 1340–1346. doi:10.1016/j.carres.2009.05.016
Return to citation in text: [1] [2] -
Yu, H. N.; Furukawa, J.-i.; Ikeda, T.; Wong, C.-H. Org. Lett. 2004, 6, 723–726. doi:10.1021/ol036390m
Return to citation in text: [1] [2] -
Agnihotri, G.; Misra, A. K. Carbohydr. Res. 2006, 341, 2420–2425. doi:10.1016/j.carres.2006.07.007
Return to citation in text: [1] -
Kim, H.-J.; Ahn, K. C.; Ma, S. J.; Gee, S. J.; Hammock, B. D. J. Agric. Food Chem. 2007, 55, 3750–3757. doi:10.1021/jf063282g
Return to citation in text: [1] -
Koenigs, W.; Knorr, E. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. doi:10.1002/cber.190103401162
Return to citation in text: [1] [2] -
Epp, J. B.; Widlanski, T. S. J. Org. Chem. 1999, 64, 293–295. doi:10.1021/jo981316g
Return to citation in text: [1] -
Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v
Return to citation in text: [1] -
Pedersen, D. S.; Rosenbohm, C. Synthesis 2001, 2431–2434. doi:10.1055/s-2001-18722
Return to citation in text: [1]
| 17. | Djerassi, C.; Sheehan, M.; Spangler, R. J. J. Org. Chem. 1971, 36, 3526–3532. doi:10.1021/jo00822a013 |
| 18. | George, S.; Sudalai, A. Tetrahedron: Asymmetry 2007, 18, 975–981. doi:10.1016/j.tetasy.2007.04.008 |
| 19. | Raunkjær, M.; Pedersen, D. S.; Elsey, G. M.; Sefton, M. A.; Skouroumounis, G. K. Tetrahedron Lett. 2001, 42, 8717–8719. doi:10.1016/S0040-4039(01)01890-1 |
| 1. | Carter, L. P.; Pardi, D.; Gorsline, J.; Griffiths, R. R. Drug Alcohol Depend. 2009, 104, 1–10. doi:10.1016/j.drugalcdep.2009.04.012 |
| 5. | Jatlow, P.; O'Malley, S. S. Alcohol.: Clin. Exp. Res. 2010, 34, 968–975. doi:10.1111/j.1530-0277.2010.01171.x |
| 21. | Brown, R. T.; Carter, N. K.; Lumbard, K. W.; Scheinmann, F. Tetrahedron Lett. 1995, 36, 8661–8664. doi:10.1016/0040-4039(95)01786-H |
| 22. | Brown, R. T.; Carter, N. E.; Mayalarp, S. P.; Scheinmann, F. Tetrahedron 2000, 56, 7591–7594. doi:10.1016/S0040-4020(00)00664-5 |
| 23. | Lucas, R.; Alcantara, D.; Morales, J. C. Carbohydr. Res. 2009, 344, 1340–1346. doi:10.1016/j.carres.2009.05.016 |
| 2. | Knudsen, K.; Greter, J.; Verdicchio, M. Clin. Toxicol. 2008, 46, 187–192. doi:10.1080/15563650701263633 |
| 14. | Bollenback, G. N.; Long, J. W.; Benjamin, D. G.; Lindquist, J. A. J. Am. Chem. Soc. 1955, 77, 3310–3315. doi:10.1021/ja01617a047 |
| 24. | Yu, H. N.; Furukawa, J.-i.; Ikeda, T.; Wong, C.-H. Org. Lett. 2004, 6, 723–726. doi:10.1021/ol036390m |
| 3. | Haller, C.; Thai, D.; Jacob, P. I.; Dyer, J. E. J. Anal. Toxicol. 2006, 30, 360–364. |
| 4. | Brailsford, A. D.; Cowan, D. A.; Kicman, A. T. J. Anal. Toxicol. 2012, 36, 88–95. doi:10.1093/jat/bkr023 |
| 11. | Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311 |
| 2. | Knudsen, K.; Greter, J.; Verdicchio, M. Clin. Toxicol. 2008, 46, 187–192. doi:10.1080/15563650701263633 |
| 20. | Berrang, B.; Brine, G. A.; Carroll, F. I. Synthesis 1997, 1165–1168. doi:10.1055/s-1997-3187 |
| 21. | Brown, R. T.; Carter, N. K.; Lumbard, K. W.; Scheinmann, F. Tetrahedron Lett. 1995, 36, 8661–8664. doi:10.1016/0040-4039(95)01786-H |
| 22. | Brown, R. T.; Carter, N. E.; Mayalarp, S. P.; Scheinmann, F. Tetrahedron 2000, 56, 7591–7594. doi:10.1016/S0040-4020(00)00664-5 |
| 23. | Lucas, R.; Alcantara, D.; Morales, J. C. Carbohydr. Res. 2009, 344, 1340–1346. doi:10.1016/j.carres.2009.05.016 |
| 11. | Schmidt, R. R.; Michel, J. Angew. Chem., Int. Ed. Engl. 1980, 19, 731–732. doi:10.1002/anie.198007311 |
| 13. | Brown, R. T.; Scheinmann, F.; Stachulski, A. V. J. Chem. Res., Synop. 1997, 370–371. |
| 14. | Bollenback, G. N.; Long, J. W.; Benjamin, D. G.; Lindquist, J. A. J. Am. Chem. Soc. 1955, 77, 3310–3315. doi:10.1021/ja01617a047 |
| 15. | Trynda, A.; Madaj, J.; Konitz, A.; Wiśniewski, A. Carbohydr. Res. 2000, 329, 249–252. doi:10.1016/S0008-6215(00)00157-9 |
| 16. | Dumont-Hornebeck, B. A.; Joly, J.-P.; Coulon, J.; Chapleur, Y. Carbohydr. Res. 1999, 320, 147–160. doi:10.1016/S0008-6215(99)00151-2 |
| 9. | Wilkinson, S. M.; Liew, C. W.; Mackay, J. P.; Salleh, H. M.; Withers, S. G.; McLeod, M. D. Org. Lett. 2008, 10, 1585–1588. doi:10.1021/ol8002767 |
| 10. | Khymenets, O.; Joglar, J.; Clapés, P.; Parella, T.; Covas, M.-I.; de la Torre, R. Adv. Synth. Catal. 2006, 348, 2155–2162. doi:10.1002/adsc.200606221 |
| 17. | Djerassi, C.; Sheehan, M.; Spangler, R. J. J. Org. Chem. 1971, 36, 3526–3532. doi:10.1021/jo00822a013 |
| 18. | George, S.; Sudalai, A. Tetrahedron: Asymmetry 2007, 18, 975–981. doi:10.1016/j.tetasy.2007.04.008 |
| 19. | Raunkjær, M.; Pedersen, D. S.; Elsey, G. M.; Sefton, M. A.; Skouroumounis, G. K. Tetrahedron Lett. 2001, 42, 8717–8719. doi:10.1016/S0040-4039(01)01890-1 |
| 7. | Stachulski, A. V.; Jenkins, G. N. Nat. Prod. Rep. 1998, 15, 173–186. doi:10.1039/a815173y |
| 8. | Kaspersen, F. M.; van Boeckel, C. A. A. Xenobiotica 1987, 17, 1451–1471. doi:10.3109/00498258709044005 |
| 6. | Petersen, I. N.; Kristensen, J. L.; Tortzen, C.; Pedersen, D. S.; Breindahl, T. J. Anal. Toxicol. 2013, in press. |
| 7. | Stachulski, A. V.; Jenkins, G. N. Nat. Prod. Rep. 1998, 15, 173–186. doi:10.1039/a815173y |
| 8. | Kaspersen, F. M.; van Boeckel, C. A. A. Xenobiotica 1987, 17, 1451–1471. doi:10.3109/00498258709044005 |
| 12. | Pews-Davtyan, A.; Pirojan, A.; Shaljyan, I.; Awetissjan, A. A.; Reinke, H.; Vogel, C. J. Carbohydr. Chem. 2003, 22, 939–962. doi:10.1081/CAR-120026603 |
| 27. | Koenigs, W.; Knorr, E. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. doi:10.1002/cber.190103401162 |
| 7. | Stachulski, A. V.; Jenkins, G. N. Nat. Prod. Rep. 1998, 15, 173–186. doi:10.1039/a815173y |
| 8. | Kaspersen, F. M.; van Boeckel, C. A. A. Xenobiotica 1987, 17, 1451–1471. doi:10.3109/00498258709044005 |
| 25. | Agnihotri, G.; Misra, A. K. Carbohydr. Res. 2006, 341, 2420–2425. doi:10.1016/j.carres.2006.07.007 |
| 26. | Kim, H.-J.; Ahn, K. C.; Ma, S. J.; Gee, S. J.; Hammock, B. D. J. Agric. Food Chem. 2007, 55, 3750–3757. doi:10.1021/jf063282g |
| 27. | Koenigs, W.; Knorr, E. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. doi:10.1002/cber.190103401162 |
| 24. | Yu, H. N.; Furukawa, J.-i.; Ikeda, T.; Wong, C.-H. Org. Lett. 2004, 6, 723–726. doi:10.1021/ol036390m |
| 13. | Brown, R. T.; Scheinmann, F.; Stachulski, A. V. J. Chem. Res., Synop. 1997, 370–371. |
| 14. | Bollenback, G. N.; Long, J. W.; Benjamin, D. G.; Lindquist, J. A. J. Am. Chem. Soc. 1955, 77, 3310–3315. doi:10.1021/ja01617a047 |
| 15. | Trynda, A.; Madaj, J.; Konitz, A.; Wiśniewski, A. Carbohydr. Res. 2000, 329, 249–252. doi:10.1016/S0008-6215(00)00157-9 |
| 16. | Dumont-Hornebeck, B. A.; Joly, J.-P.; Coulon, J.; Chapleur, Y. Carbohydr. Res. 1999, 320, 147–160. doi:10.1016/S0008-6215(99)00151-2 |
| 29. | Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v |
| 30. | Pedersen, D. S.; Rosenbohm, C. Synthesis 2001, 2431–2434. doi:10.1055/s-2001-18722 |
| 6. | Petersen, I. N.; Kristensen, J. L.; Tortzen, C.; Pedersen, D. S.; Breindahl, T. J. Anal. Toxicol. 2013, in press. |
| 6. | Petersen, I. N.; Kristensen, J. L.; Tortzen, C.; Pedersen, D. S.; Breindahl, T. J. Anal. Toxicol. 2013, in press. |
| 19. | Raunkjær, M.; Pedersen, D. S.; Elsey, G. M.; Sefton, M. A.; Skouroumounis, G. K. Tetrahedron Lett. 2001, 42, 8717–8719. doi:10.1016/S0040-4039(01)01890-1 |
| 28. | Epp, J. B.; Widlanski, T. S. J. Org. Chem. 1999, 64, 293–295. doi:10.1021/jo981316g |
© 2013 Nymann Petersen et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)