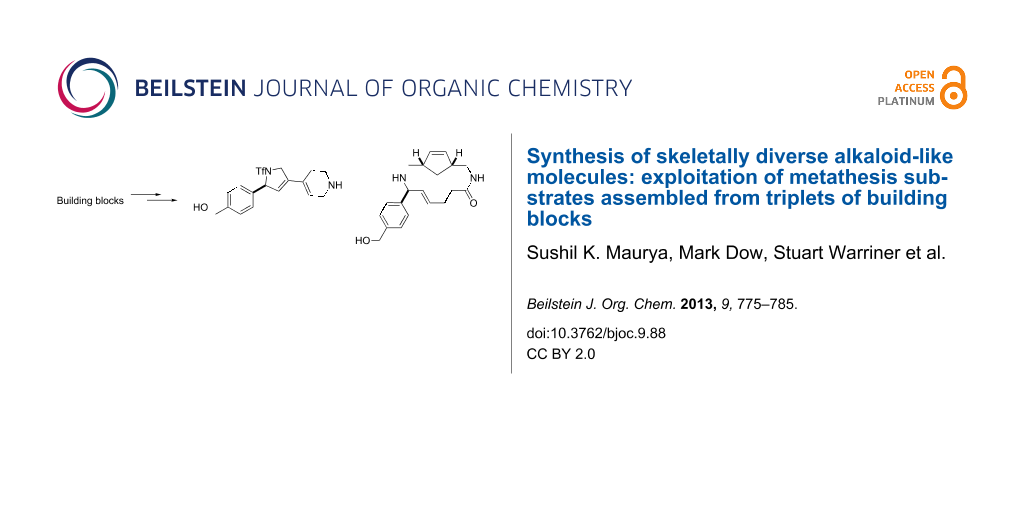
Abstract
A range of metathesis substrates was assembled from triplets of unsaturated building blocks. The approach involved the iterative attachment of a propagating and a terminating building block to a fluorous-tagged initiating building block. Metathesis cascade chemistry was used to “reprogram” the molecular scaffolds. Remarkably, in one case, a cyclopropanation reaction competed with the expected metathesis cascade process. Finally, it was demonstrated that the metathesis products could be derivatised to yield the final products. At each stage, purification was facilitated by the presence of a fluorous-tagged protecting group.

Graphical Abstract
Introduction
Our collective understanding of the biological relevance of chemical space has been shaped, in large part, by the historic exploration of chemical space by chemical synthesis (and biosynthesis) [1]. The scaffolds of known bioactive small molecules, in particular, play a key role in guiding the navigation of chemical space [2-4]. The field of biology-oriented synthesis (BIOS) [5], for example, uses biologically validated scaffolds [6-8] to inspire library design.
Known organic molecules populate chemical space unevenly and unsystematically. Around half of all known organic compounds are based on only 0.25% of the known molecular scaffolds [9]! This uneven coverage of chemical space is also typical of small-molecule screening collections [7,10]. Consequently, the biological relevance of most known scaffolds has been poorly explored. The field of diversity-oriented synthesis [11-13] has emerged with the specific aim of populating screening collections with diverse and novel small molecules.
We have previously developed a robust approach for the synthesis of skeletally diverse small molecules (Scheme 1) [14]. The approach relied on the synthesis of metathesis substrates by iterative attachment of simple unsaturated building blocks to a fluorous-tagged linker 1 (e.g., → 2 or 3). Subsequently, metathesis cascade reactions were used to “reprogram” the molecular scaffolds, concomitantly releasing the products from the linker (e.g., → 4 or 5) [14-17]. The approach enabled the combinatorial variation of molecular scaffolds, and was exploited in the synthesis of natural-product-like small molecules with unprecedented scaffold diversity (over 80 distinct scaffolds).
![[1860-5397-9-88-i1]](/bjoc/content/inline/1860-5397-9-88-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Illustrative examples of a synthetic approach to natural-product-like molecules with over eighty molecular scaffolds.
Scheme 1: Illustrative examples of a synthetic approach to natural-product-like molecules with over eighty mo...
Although powerful, this general approach to skeletally diverse molecules had only been exemplified by varying pairs of unsaturated building blocks [14]. Thus, by exploiting the linker 1, which is an allyl alcohol or allyl amine equivalent, all of the products were inevitably allylic alcohols or cyclic allylic amines. Here, we demonstrate that the approach is considerably more general, and that it is feasible to exploit triplets of building blocks, extending the range of diverse molecular scaffolds that may be prepared.
Results and Discussion
Library design
An overview of the proposed approach to the synthesis of diverse scaffolds is shown in Scheme 2. The building blocks used in this study are shown in Figure 1. It was planned to start with an “initiating” building block (e.g., 6a or 7) bearing a fluorous tag to facilitate the purification of synthetic intermediates [18]. Iterative attachment of a propagating and a terminating building block would yield a metathesis substrate (such as 14 or 16). Finally, a metathesis cascade reaction would yield a product scaffold (such as 15 and 17). It was planned that many of the product scaffolds would bear an o-nitrophenylsulfonyl protecting group. The combinations of building blocks were carefully chosen to ensure that, after deprotection, selective derivatisation of the product scaffolds would be possible.
![[1860-5397-9-88-i2]](/bjoc/content/inline/1860-5397-9-88-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Overview of the proposed synthetic approach. FDIPES = diisopropyl(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)silyl; Ns = o-nitrophenylsulfonyl.
Scheme 2: Overview of the proposed synthetic approach. FDIPES = diisopropyl(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10...
![[1860-5397-9-88-1]](/bjoc/content/figures/1860-5397-9-88-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of building blocks used in this study. Panel A: fluorous-tagged initiating building blocks. Panel B: propagating building blocks. Panel C: terminating building blocks.
Figure 1: Structures of building blocks used in this study. Panel A: fluorous-tagged initiating building bloc...
Synthesis of building blocks
The initiating building blocks 6a and 6b were prepared by using the approach outlined in Scheme 3. The allylic alcohol 14 [19] was converted into the allylic carbonate 15 by treatment with methyl chloroformate and DMAP. The allylic carbonate 15 underwent efficient asymmetric allylic amination [20] with o-nitrophenylsulfonamide as the nucleophile to give the allylic sulfonamide 17 in 66% yield; in addition, the linear product 16 was also obtained in 7% yield. Desilylation of 17 (→ 18) and reaction with diisopropyl(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)silyl (FDIPES) bromide, generated in situ from the corresponding silyl hydride, gave the fluorous-tagged building block 6b. Finally, desulfonylation (→ 19) and trifluoromethylsulfonylation yielded the alternative initiating building block 6a.
![[1860-5397-9-88-i3]](/bjoc/content/inline/1860-5397-9-88-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the initiating building blocks 6a and 6b. TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene.
Scheme 3: Synthesis of the initiating building blocks 6a and 6b. TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene.
The initiating building block 7 was prepared from the sulfinimine 21 by adapting a synthesis previously reported by Ellman (Scheme 4) [21]. Treatment of the sulfinimine 21 in dichloromethane with allylmagnesium bromide yielded the corresponding sulfinimides as a 79:21 mixture of diastereoiomers; following column chromatography, the major diastereomer 22 was obtained in 70% yield, and was converted into the corresponding amino alcohol 23. The configuration of the amino alcohol 23 was determined by conversion into the corresponding benzamide and comparison with racemic and enantiomerically enriched samples (prepared from the commercially available amino acid). Analysis by chiral HPLC indicated that the amino alcohol 23 had (R)-configuration. It was concluded that the sense of diastereoselectivity in the addition 21 → 22 contrasted with that reported by Ellman [21]. However, the sense of diastereoselectivity was the same as that reported for the addition of allylmagnesium bromide in dichloromethane to a similar sulfinimine [22]. The amino alcohol 23 was converted into the corresponding o-nitrophenylsulfonamide 24 and, hence, the fluorous-tagged building block 7.
![[1860-5397-9-88-i4]](/bjoc/content/inline/1860-5397-9-88-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the initiating building block 7.
Scheme 4: Synthesis of the initiating building block 7.
The propagating building blocks 8–11, and the terminating building block 12b, were prepared by using established methods [14]. The enantiomeric excess (68% ee) of the hydroxy alcohol 11 was determined by conversion into the corresponding diastereomeric O-methyl mandelate esters. The terminating building blocks 12a and 13 were prepared by straightforward derivatisation of commercially available starting materials (see Supporting Information File 1).
Synthesis of metathesis substrates
Initially, the propagating building blocks 8–11 were attached to the fluorous-tagged initiating building blocks (6a, 6b or 7). In each case, an excess of the propagating building block, DEAD and triphenylphosphine was used. In general, the crude product was directly deacetylated. At each stage, the required fluorous-tagged product was isolated by fluorous-solid-phase extraction (F-SPE), and its purity determined by analysis by 500 MHz 1H NMR spectroscopy. These results are summarised in Table 1.
Table 1: Attachment of propagating building blocks to the fluorous-tagged initiating building blocks.
| Building blocks | Attachment | Deacetylationa | Product |
|---|---|---|---|
|
Methoda (mass recovery / %)
{Purityb / %} |
Mass recovery / %
(Purityb / %) |
||
| 6a, 8 | A1 (70) {>98} | 92 (92) |
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-88-i6.svg?max-width=637&scale=1.0)
25 |
| 6a, 9 | A2 (97) {92} | 87 (93) |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-88-i7.svg?max-width=637&scale=1.0)
26 |
| 6a, 10c | A3 (85) {>98} | 87 (98) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-88-i8.svg?max-width=637&scale=1.0)
27 |
| 6b, 11d | A3 (85) {76} | 85e (72) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-88-i9.svg?max-width=637&scale=1.0)
28 |
| 7, 8 | A3 (92) {91} | 94f |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-88-i10.svg?max-width=637&scale=1.0)
29 |
| 7, 9 | A3 (74f) | 97 (98) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-88-i11.svg?max-width=637&scale=1.0)
30 |
| 7, 10 | A3 (97) {91} | 80f |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-88-i12.svg?max-width=637&scale=1.0)
31 |
aMethods: A1: Initiating building block (1.0 equiv), propagating building block (4.0 equiv), DEAD (4.0 equiv), PPh3 (4.0 equiv), CH2Cl2, 0 °C → rt then F–SPE; A2: Initiating building block (1.0 equiv), propagating building block (4.0 equiv), DEAD (2.0 equiv), PPh3 (2.0 equiv), CH2Cl2, 0 °C → rt then F–SPE; A3: Initiating building block (1.0 equiv), propagating building block (4.0 equiv), DEAD (2.0 equiv), PPh3 (2.0 equiv), THF, 0 °C → rt then F–SPE; Deacetylation: 0.025 M NH3 in MeOH. bDetermined by analysis of the 500 MHz 1H NMR spectrum. cThe building block had >98% ee. dThe building block had 68% ee. eIsolated as a ca. 75:25 mixture of diastereoisomers. fIsolated yield of purified product (see Supporting Information File 1).
The metathesis substrates were prepared by subsequent attachment of a terminating building block (12a, 12b or 13) (see Table 2). In each case, an excess of the terminating building block, DEAD and triphenylphosphine was used; the required fluorous-tagged product was isolated by solid-fluorous phase extraction (F-SPE), and its purity was determined by analysis by 500 MHz 1H NMR spectroscopy.
Table 2: Attachment of propagating building blocks to the fluorous-tagged initiating building blocks.
| Substrate | Terminating building block | Attachment | Product |
|---|---|---|---|
|
Methoda (mass recovery / %)
{Purityb / %} |
|||
| 25 | 12b | A4 (89) {83} |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-88-i13.svg?max-width=637&scale=1.0)
32 |
| 25 | 13 | A4 (89) {86} |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-88-i14.svg?max-width=637&scale=1.0)
33 |
| 26 | 12b | A4 (76) {93}] |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-88-i15.svg?max-width=637&scale=1.0)
34 |
| 26 | 13 | A4 (75) {97} |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-88-i16.svg?max-width=637&scale=1.0)
35 |
| 27 | 12b | A5 (62c) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-88-i17.svg?max-width=637&scale=1.0)
36 |
| 27 | 13 | A5 (54c) |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-88-i18.svg?max-width=637&scale=1.0)
37 |
| 28d | 12a | A5 (86c,e) |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-88-i19.svg?max-width=637&scale=1.0)
38 |
| 28d | 13 | A5 (77c,e) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-88-i20.svg?max-width=637&scale=1.0)
39 |
| 29 | 12a | A6 (86c) |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-88-i21.svg?max-width=637&scale=1.0)
40 |
| 29 | 13 | A6 (77c) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-88-i22.svg?max-width=637&scale=1.0)
41 |
| 30 | 12a | A6 (92c) |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-88-i23.svg?max-width=637&scale=1.0)
42 |
| 30 | 13 | A6 (55c) |
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-88-i24.svg?max-width=637&scale=1.0)
43 |
| 31 | 12a | A6 (85c) |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-88-i25.svg?max-width=637&scale=1.0)
44 |
aMethods: A4: Substrate (1.0 equiv), propagating building block (4.0 equiv), DEAD (2.0 equiv), PPh3 (2.0 equiv), CH2Cl2, 0 °C → rt then F-SPE; A5: Substrate (1.0 equiv), propagating building block (4.0 equiv), DEAD (4.0 equiv), PPh3 (4.0 equiv), THF, 0 °C → rt then F-SPE; A6: Substrate (1.0 equiv), propagating building block (4.0 equiv), DEAD (2.0 equiv), PPh3 (2.0 equiv), THF, 0 °C → rt then F-SPE. bDetermined by analysis of the 500 MHz 1H NMR spectrum. cIsolated yield of purified product. dThe starting material was a ca. 75:25 mixture of diastereoisomers. eIsolated as a ca. 75:25 mixture of diastereomers.
Metathesis cascade reactions
The scaffolds of the metathesis substrates were “reprogrammed” by treatment with Hoveyda–Grubbs second-generation catalyst in either dichloromethane or tert-butyl methyl ether [23] (TBME). Many of the metathesis reactions were rather sluggish, and the catalyst was added portionwise until the reactions were judged to be complete by TLC analysis. After removal [24] of the catalyst by using tris(hydroxymethyl)phosphine, the metathesis products were generally purified by flash column chromatography. Finally, the o-nitrophenylsulfonyl groups were removed from the products. The results are summarised in Table 3.
Table 3: Application of cascade metathesis reactions in the synthesis of diverse scaffolds and subsequent desulfonylation.
| Substrate | Methoda (mol %; time) | Product | Yield / % |
|---|---|---|---|
| 32 | B1 (5 + 2.5; 3 d) then C1 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-88-i26.svg?max-width=637&scale=1.0)
45 |
45, 37%b |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-88-i27.svg?max-width=637&scale=1.0)
46 |
46, 11%b | ||
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-88-i28.svg?max-width=637&scale=1.0)
47 |
47, 5%b | ||
| 33 | B1 (2 × 5; 4 d) then C1 then D |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-88-i29.svg?max-width=637&scale=1.0)
48 |
43%b |
| 34 | B1 (5 + 5 + 2.5; 10 d) then C1 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-88-i30.svg?max-width=637&scale=1.0)
49 |
49%b (86%c) |
| 36 | B1 (4 × 5; 20 d) then C1 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-88-i31.svg?max-width=637&scale=1.0)
50 |
77 then 81 (93%c) |
| 38d | B1 (3 × 5; 14 d) then C1 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-88-i32.svg?max-width=637&scale=1.0)
51 |
63 then 85 (51) |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-88-i33.svg?max-width=637&scale=1.0)
52 |
29 then 94 (52) | ||
| 39 | B1 (4 × 5; 20 d) then C2 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-88-i34.svg?max-width=637&scale=1.0)
53 |
8%b |
| 40 | B2 (5; 24 h) then C1 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-9-88-i35.svg?max-width=637&scale=1.0)
54 |
93 then 96 (87%c) |
| 41 | B2 (5; 24 h) then C2 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-9-88-i36.svg?max-width=637&scale=1.0)
55 |
93 then 80 (93%c) |
| 42 | B2 (3 × 5; 7 d) then C1 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-9-88-i37.svg?max-width=637&scale=1.0)
56 |
76 then 92 (92%c) |
| 43 | B2 (2 × 5; 3 d) then C2 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-9-88-i38.svg?max-width=637&scale=1.0)
57 |
54 then 77 (86%c) |
| 44 | B2 (5; 24 h) then C1 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-9-88-i39.svg?max-width=637&scale=1.0)
58 |
53 then 99 (98%c) |
aMethods: B1: Hoveyda–Grubbs second-generation catalyst, CH2Cl2, 50 °C then Et3N (86 equiv), P(CH2OH)3 (86 equiv) then silica; B2: Hoveyda–Grubbs second-generation catalyst, MTBE, 50 °C then Et3N (86 equiv), P(CH2OH)3 (86 equiv) then silica; C1: PhSH (1.2 equiv), K2CO3 (3.0 equiv), DMF; C2: PhSH (2.4 equiv), K2CO3 (6.0 equiv), DMF; E: aq HF, MeCN–CH2Cl2. bYield over more than one step. cPurity of the product determined by 500 MHz 1H NMR spectroscopy. dThe starting material was a ca. 75:25 mixture of diastereoisomers.
In general, the metathesis reactions proceeded smoothly to give the expected metathesis cascade products. In the case of 39, however, the cyclopentene did not participate in the metathesis reaction, and the bridged macrocycle 53 was obtained in low yield. We have previously observed the formation of macrocyclic metathesis products in similar metathesis cascade reactions [14]. The formation of the cyclopropanes 46 and 47 as byproducts in the metathesis cascade reaction of 32 was remarkable [25]. Presumably, in this case, the metathesis cascade leads to the generation of the intermediate 59 (Scheme 5); the intermediate could then react to conclude the metathesis cascade (to give 45 after deprotection), or cyclopropanate [25] the terminal alkene (to give 46 or 47 after deprotection) (Scheme 5).
![[1860-5397-9-88-i5]](/bjoc/content/inline/1860-5397-9-88-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Fate of the metathesis reaction of the substrate 32.
Scheme 5: Fate of the metathesis reaction of the substrate 32.
Finally, a selection of fluorous-tagged products was derivatised (typically on a 50 μmol scale) to yield a range of amides and ureas (Table 4). The fluorous tag facilitated the purification of the derivitised products by F-SPE. The final products 60–71 (Table 4) were obtained after removal of the fluorous tag by desilylation.
Table 4: Derivatisation and deprotection of final products.
| Substrate (puritya / %) | Productb | Methodc | Yield / % | |
|---|---|---|---|---|
| 45 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-9-88-i40.svg?max-width=637&scale=1.0)
|
60a | D | 51 |
| 60b | E1 then D | 81 then 60 | ||
| 60c | E2 then D | 67 then 98 | ||
| 60d | E3 then D | 94 then 81 | ||
| 46 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-9-88-i41.svg?max-width=637&scale=1.0)
|
61b | E1 then D | 39 then 70 |
| 47 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-9-88-i42.svg?max-width=637&scale=1.0)
|
62b | E1 then D | 43 then 63 |
|
49
(86) |
![[Graphic 38]](/bjoc/content/inline/1860-5397-9-88-i43.svg?max-width=637&scale=1.0)
|
63a | D | 83 |
| 63b | E1 then D | 32 then 64 | ||
| 63c | E2 then D | 83 then 58 | ||
| 63d | E3 then D | 84 then 79 | ||
|
50
(93) |
![[Graphic 39]](/bjoc/content/inline/1860-5397-9-88-i44.svg?max-width=637&scale=1.0)
|
64a | D | 87 |
| 64b | E1 then D | 29d | ||
| 64c | E2 then D | 43d | ||
| 64d | E3 then D | 34d | ||
|
51
(85) |
![[Graphic 40]](/bjoc/content/inline/1860-5397-9-88-i45.svg?max-width=637&scale=1.0)
|
65a | D | 70 |
| 65b | E1 then D | 40d | ||
| 65c | E2 then D | 82d | ||
| 65d | E3 then D | 74d | ||
| 53 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-9-88-i46.svg?max-width=637&scale=1.0)
|
66a | D | 52 |
|
54
(87) |
![[Graphic 42]](/bjoc/content/inline/1860-5397-9-88-i47.svg?max-width=637&scale=1.0)
|
67a | D | 91 |
| 67b | E4 then D | 77d | ||
| 67c | E2 then D | 83d | ||
| 67d | E3 then D | 42d | ||
|
55
(93) |
![[Graphic 43]](/bjoc/content/inline/1860-5397-9-88-i48.svg?max-width=637&scale=1.0)
|
68a | D | 94 |
| 68c | E2 then D | 67d | ||
| 68d | E3 then D | 40d | ||
|
56
(92) |
![[Graphic 44]](/bjoc/content/inline/1860-5397-9-88-i49.svg?max-width=637&scale=1.0)
|
69a | D | 91 |
| 69b | E4 then D | 67d | ||
| 69c | E2 then D | 67d | ||
|
57
(86) |
![[Graphic 45]](/bjoc/content/inline/1860-5397-9-88-i50.svg?max-width=637&scale=1.0)
|
70a | D | 47 |
| 70c | E2 then D | 59d | ||
|
58
(98) |
![[Graphic 46]](/bjoc/content/inline/1860-5397-9-88-i51.svg?max-width=637&scale=1.0)
|
71a | D | 91 |
| 71b | E4 then D | 64d | ||
| 71c | E2 then D | 53d | ||
| 71d | E3 then D | 29d | ||
aDetermined by analysis of the product by 500 MHz 1H NMR spectroscopy. bThe suffix refers to the identity of the R substituent: a, R = H; b, R = isoxazole-5-carbonyl; c, R = pyridine-3-carbonylamino; d, R = morpholine-4-carbonyl; cMethods: D: aq HF, MeCN–CH2Cl2; E1: isoxazole-5-carbonyl chloride (2.0 equiv), Et3N (3.0 equiv), DMAP (1.0 equiv), CH2Cl2; E2: pyridine-3-isocyanate (2.0 equiv), Et3N (3.0 equiv), DMAP (1.0 equiv), CH2Cl2; E3: morpholine-4-carbonyl chloride (2.0 equiv), Et3N (3.0 equiv), DMAP (1.0 equiv), CH2Cl2; E4: isoxazole-5-carbonyl chloride (2.0 equiv), pyridine; dYield over two steps.
Conclusion
Metathesis is an extremely powerful reaction for diversity-oriented synthesis. It was demonstrated that metathesis substrates could be assembled efficiently from triplets of building blocks. Thereafter, metathesis cascades yielded a diverse range of molecular scaffolds. The diversity of the products was increased through variation of all three of the building blocks used: the initiating, the propagating, and the terminating building block.
The overall approach was facilitated by fluorous tagging of the initiating building block, allowing easy purification (by F-SPE) of synthetic intermediates and metathesis products. The presence of a fluorous tag also facilitated the purification of the functionalised products. Evaluation of the biological activity of the final products will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental and compound characterisation. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Paolini, G. V.; Shapland, R. H. B.; van Hoorn, W. P.; Mason, J. S.; Hopkins, A. L. Nat. Biotechnol. 2006, 24, 805–815. doi:10.1038/nbt1228
Return to citation in text: [1] -
Wetzel, S.; Klein, K.; Renner, S.; Rauh, D.; Oprea, T. I.; Mutzel, P.; Waldmann, H. Nat. Chem. Biol. 2009, 5, 581–583. doi:10.1038/nchembio.187
Return to citation in text: [1] -
Renner, S.; van Otterlo, W. A. L.; Dominguez Seoane, M.; Möcklinghoff, S.; Hofmann, B.; Wetzel, S.; Schuffenhauer, A.; Ertl, P.; Oprea, T. I.; Steinhilber, D.; Brunsveld, L.; Rauh, D.; Waldmann, H. Nat. Chem. Biol. 2009, 5, 585–592. doi:10.1038/nchembio.188
Return to citation in text: [1] -
Hu, Y.; Wassermann, A. M.; Lounkine, E.; Bajorath, J. J. Med. Chem. 2010, 53, 752–758. doi:10.1021/jm9014229
Return to citation in text: [1] -
Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800–10826. doi:10.1002/anie.201007004
Return to citation in text: [1] -
Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002
Return to citation in text: [1] -
Shelat, A. A.; Guy, R. K. Nat. Chem. Biol. 2007, 3, 442–446. doi:10.1038/nchembio0807-442
Return to citation in text: [1] [2] -
Hu, Y.; Stumpfe, D.; Bajorath, J. J. Chem. Inf. Model. 2011, 51, 1742–1753. doi:10.1021/ci200179y
Return to citation in text: [1] -
Lipkus, A. H.; Yuan, Q.; Lucas, K. A.; Funk, S. A.; Bartelt, W. F., III; Schenck, R. J.; Trippe, A. J. J. Org. Chem. 2008, 73, 4443–4451. doi:10.1021/jo8001276
Return to citation in text: [1] -
Krier, M.; Bret, G.; Rognan, D. J. Chem. Inf. Model. 2006, 46, 512–524. doi:10.1021/ci050352v
Return to citation in text: [1] -
O' Connor, C. J.; Beckmann, H. S. G.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 4444–4456. doi:10.1039/c2cs35023h
See for a recent review of diversity-oriented synthesis.
Return to citation in text: [1] -
MacLellan, P.; Nelson, A. Chem. Commun. 2013, 49, 2383–2393. doi:10.1039/c2cc38184b
See for a recent review of diversity-oriented synthesis.
Return to citation in text: [1] -
Nielsen, T. E.; Schreiber, S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. doi:10.1002/anie.200703073
See for a recent review of diversity-oriented synthesis.
Return to citation in text: [1] -
Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486
Return to citation in text: [1] [2] [3] [4] [5] -
Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/b807278g
See for other diversity-oriented approaches that exploit metathesis chemistry.
Return to citation in text: [1] -
Spiegel, D. A.; Schroeder, F. C.; Duvall, J. R.; Schreiber, S. L. J. Am. Chem. Soc. 2006, 128, 14766–14767. doi:10.1021/ja065724a
See for other diversity-oriented approaches that exploit metathesis chemistry.
Return to citation in text: [1] -
O'Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563–9571. doi:10.1002/chem.201000707
See for other diversity-oriented approaches that exploit metathesis chemistry.
Return to citation in text: [1] -
Gladysz, A.; Curran, D. P.; Horváth, I. T. Handbook of Fluorous Chemistry; Wiley: New York, 2004. doi:10.1002/3527603905
Return to citation in text: [1] -
Wuts, P. G. M.; Ashford, S. W.; Anderson, A. M.; Atkins, J. R. Org. Lett. 2003, 5, 1483–1485. doi:10.1021/ol034274d
Return to citation in text: [1] -
Weinhofen, R.; Tverskoy, O.; Helmchen, G. Angew. Chem., Int. Ed. 2006, 45, 5546–5549. doi:10.1002/anie.200601472
Return to citation in text: [1] -
Rech, J. C.; Yato, M.; Duckett, D.; Ember, B.; LoGrosso, P. V.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 490–491. doi:10.1021/ja0676004
Return to citation in text: [1] [2] -
Koriyama, Y.; Nozawa, A.; Hayakawa, R.; Shimizu, M. Tetrahedron 2002, 58, 9621–9628. doi:10.1016/S0040-4020(02)01250-4
Return to citation in text: [1] -
Kuhn, K. M.; Champagne, T. M.; Hong, S. H.; Wei, W.-H.; Nickel, A.; Lee, C. W.; Virgil, S. C.; Grubbs, R. H.; Pederson, R. L. Org. Lett. 2010, 12, 984–987. doi:10.1021/ol9029808
Return to citation in text: [1] -
Maynard, H. D.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 4137–4140. doi:10.1016/S0040-4039(99)00726-1
Return to citation in text: [1] -
Peppers, B. P.; Diver, S. T. J. Am. Chem. Soc. 2004, 126, 9524–9525. doi:10.1021/ja049079o
See for a reaction with a similar outcome.
Return to citation in text: [1] [2]
| 24. | Maynard, H. D.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 4137–4140. doi:10.1016/S0040-4039(99)00726-1 |
| 14. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 23. | Kuhn, K. M.; Champagne, T. M.; Hong, S. H.; Wei, W.-H.; Nickel, A.; Lee, C. W.; Virgil, S. C.; Grubbs, R. H.; Pederson, R. L. Org. Lett. 2010, 12, 984–987. doi:10.1021/ol9029808 |
| 1. | Paolini, G. V.; Shapland, R. H. B.; van Hoorn, W. P.; Mason, J. S.; Hopkins, A. L. Nat. Biotechnol. 2006, 24, 805–815. doi:10.1038/nbt1228 |
| 9. | Lipkus, A. H.; Yuan, Q.; Lucas, K. A.; Funk, S. A.; Bartelt, W. F., III; Schenck, R. J.; Trippe, A. J. J. Org. Chem. 2008, 73, 4443–4451. doi:10.1021/jo8001276 |
| 21. | Rech, J. C.; Yato, M.; Duckett, D.; Ember, B.; LoGrosso, P. V.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 490–491. doi:10.1021/ja0676004 |
| 6. | Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P. J. Med. Chem. 1988, 31, 2235–2246. doi:10.1021/jm00120a002 |
| 7. | Shelat, A. A.; Guy, R. K. Nat. Chem. Biol. 2007, 3, 442–446. doi:10.1038/nchembio0807-442 |
| 8. | Hu, Y.; Stumpfe, D.; Bajorath, J. J. Chem. Inf. Model. 2011, 51, 1742–1753. doi:10.1021/ci200179y |
| 22. | Koriyama, Y.; Nozawa, A.; Hayakawa, R.; Shimizu, M. Tetrahedron 2002, 58, 9621–9628. doi:10.1016/S0040-4020(02)01250-4 |
| 5. | Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800–10826. doi:10.1002/anie.201007004 |
| 20. | Weinhofen, R.; Tverskoy, O.; Helmchen, G. Angew. Chem., Int. Ed. 2006, 45, 5546–5549. doi:10.1002/anie.200601472 |
| 2. | Wetzel, S.; Klein, K.; Renner, S.; Rauh, D.; Oprea, T. I.; Mutzel, P.; Waldmann, H. Nat. Chem. Biol. 2009, 5, 581–583. doi:10.1038/nchembio.187 |
| 3. | Renner, S.; van Otterlo, W. A. L.; Dominguez Seoane, M.; Möcklinghoff, S.; Hofmann, B.; Wetzel, S.; Schuffenhauer, A.; Ertl, P.; Oprea, T. I.; Steinhilber, D.; Brunsveld, L.; Rauh, D.; Waldmann, H. Nat. Chem. Biol. 2009, 5, 585–592. doi:10.1038/nchembio.188 |
| 4. | Hu, Y.; Wassermann, A. M.; Lounkine, E.; Bajorath, J. J. Med. Chem. 2010, 53, 752–758. doi:10.1021/jm9014229 |
| 21. | Rech, J. C.; Yato, M.; Duckett, D.; Ember, B.; LoGrosso, P. V.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 490–491. doi:10.1021/ja0676004 |
| 14. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 15. |
Spandl, R. J.; Rudyk, H.; Spring, D. R. Chem. Commun. 2008, 3001–3003. doi:10.1039/b807278g
See for other diversity-oriented approaches that exploit metathesis chemistry. |
| 16. |
Spiegel, D. A.; Schroeder, F. C.; Duvall, J. R.; Schreiber, S. L. J. Am. Chem. Soc. 2006, 128, 14766–14767. doi:10.1021/ja065724a
See for other diversity-oriented approaches that exploit metathesis chemistry. |
| 17. |
O'Leary-Steele, C.; Pedersen, P. J.; James, T.; Lanyon-Hogg, T.; Leach, S.; Hayes, J.; Nelson, A. Chem.–Eur. J. 2010, 16, 9563–9571. doi:10.1002/chem.201000707
See for other diversity-oriented approaches that exploit metathesis chemistry. |
| 18. | Gladysz, A.; Curran, D. P.; Horváth, I. T. Handbook of Fluorous Chemistry; Wiley: New York, 2004. doi:10.1002/3527603905 |
| 25. |
Peppers, B. P.; Diver, S. T. J. Am. Chem. Soc. 2004, 126, 9524–9525. doi:10.1021/ja049079o
See for a reaction with a similar outcome. |
| 14. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 19. | Wuts, P. G. M.; Ashford, S. W.; Anderson, A. M.; Atkins, J. R. Org. Lett. 2003, 5, 1483–1485. doi:10.1021/ol034274d |
| 11. |
O' Connor, C. J.; Beckmann, H. S. G.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 4444–4456. doi:10.1039/c2cs35023h
See for a recent review of diversity-oriented synthesis. |
| 12. |
MacLellan, P.; Nelson, A. Chem. Commun. 2013, 49, 2383–2393. doi:10.1039/c2cc38184b
See for a recent review of diversity-oriented synthesis. |
| 13. |
Nielsen, T. E.; Schreiber, S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. doi:10.1002/anie.200703073
See for a recent review of diversity-oriented synthesis. |
| 14. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 7. | Shelat, A. A.; Guy, R. K. Nat. Chem. Biol. 2007, 3, 442–446. doi:10.1038/nchembio0807-442 |
| 10. | Krier, M.; Bret, G.; Rognan, D. J. Chem. Inf. Model. 2006, 46, 512–524. doi:10.1021/ci050352v |
| 14. | Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Angew. Chem., Int. Ed. 2009, 48, 104–109. doi:10.1002/anie.200804486 |
| 25. |
Peppers, B. P.; Diver, S. T. J. Am. Chem. Soc. 2004, 126, 9524–9525. doi:10.1021/ja049079o
See for a reaction with a similar outcome. |
© 2013 Maurya et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)