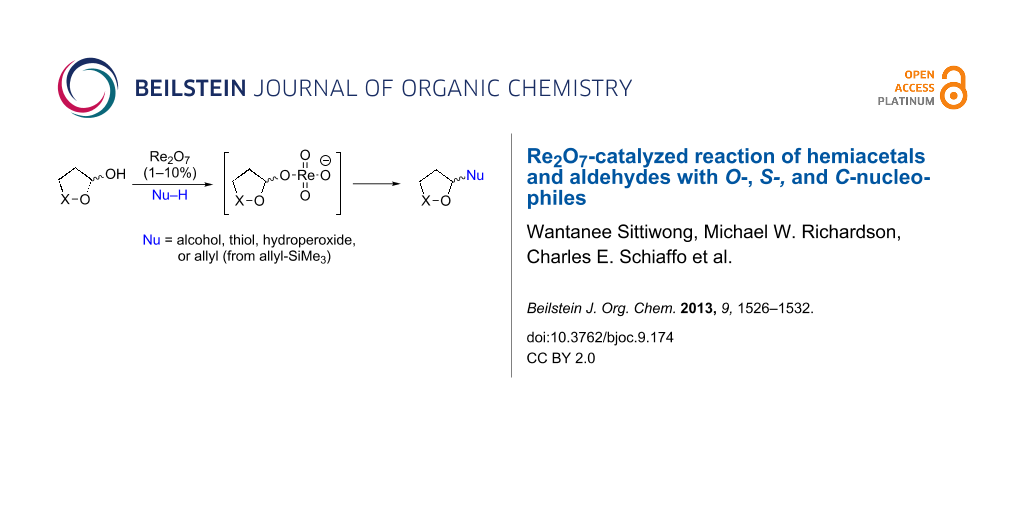
Abstract
Re(VII) oxides catalyze the acetalization, monoperoxyacetalization, monothioacetalization and allylation of hemiacetals. The reactions, which take place under mild conditions and at low catalyst loadings, can be conducted using hemiacetals, the corresponding O-silyl ethers, and, in some cases, the acetal dimers. Aldehydes react under similar conditions to furnish good yields of dithioacetals. Reactions of hemiacetals with nitrogen nucleophiles are unsuccessful. 1,2-Dioxolan-3-ols (peroxyhemiacetals) undergo Re(VII)-promoted etherification but not allylation. Hydroperoxyacetals (1-alkoxyhydroperoxides) undergo selective exchange of the alkoxide group in the presence of either Re2O7 or a Brønsted acid.

Graphical Abstract
Introduction
The synthetically important conversions of hemiacetals to acetals, thioacetals, or homoallyl ethers are typically achieved through activation of the substrate with a strong Brønsted or Lewis acid, or by conversion to an activated intermediate such as a haloacetal [1]. Perrhenates, best known as catalysts for large-scale alkene metathesis [2] and isomerization of allylic alcohols [3-10], have been shown to promote condensation of carbonyls with hydrogen peroxide or hydroperoxides [11,12], intramolecular displacements of reversibly formed hemiacetals and allylic alcohols [8,13], displacement of resonance-activated alcohols with electron-poor nitrogen nucleophiles [14], and a synthesis of homoallylated amines from condensation of carbonyl groups with an electron-poor amine in the presence of allyltrimethylsilane [15]. We now describe the Re2O7-promoted reactions of peroxyhemiacetals, hemiacetals, and alkoxyhydroperoxides with O-, S- and C-nucleophiles.
Results
In the course of investigations into potential antischistosomal and antimalarial agents [16,17], we needed to prepare a number of 3-alkoxy-1,2-dioxolanes. Brønsted acid-promoted etherification of hemiacetals (1,2-dioxolan-3-ols) required harsh conditions and proceeded in good yields only for unhindered alcohols [18]. We were curious whether Re2O7, a catalyst known to activate alcohols via a reversibly formed Re(VII) ester [19,20], would enable displacement under milder conditions, potentially allowing access to a broader range of alkoxydioxolanes. As shown in Table 1, the use of Re2O7 or p-toluenesulfonic acid monohydrate (PTSA) as catalysts provided comparable yields in the etherification of dioxolanol 1a or the corresponding O-trimethylsilyl ether 1b with an unhindered alcohol, although the perrhenate-catalyzed reaction proceeded much more rapidly. In the case of a neopentyl alcohol nucleophile, the perrhenate-catalyzed process was clearly superior, proceeding more rapidly and furnishing a higher yield of acetal. The Re2O7-promoted reactions were subsequently found to proceed efficiently at only 1% catalyst loading. Neither catalyst allowed etherification with a tertiary alcohol.
Table 1: Acetalization of hydroxy- and silyloxydioxolanes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-174-i4.svg?max-width=637&scale=1.0)
|
|||||
| R | substrate | product | catalyst (equiv) | ||
|---|---|---|---|---|---|
| PTSA (0.1) | Re2O7 (0.1) | Re2O7 (0.01) | |||
| yield (reaction time) | |||||
| Ph(CH2)2 | 1a | 2a | 73% (3 h) | 75% (1 h) | 83% (1 h) |
| 1b | 2a | — | — | 81% (2 h) | |
| 1-AdCH2a | 1a | 2b | 33% (12 h) | 74% (1 h) | 78% (2 h) |
| Ada | 1a | — | 0% | 0% | 0% |
| 1b | — | — | — | 0% | |
aAd = adamantyl.
Acetal formation
As illustrated in Table 2, we next investigated acetalization of tetrahydrofuranol 3, tetrahydropyranol 4a and the O-t-butyldimethylsilyl ether of the latter (4b). While good yields of acetals were obtained from the reaction with primary or secondary alcohols, or t-butyl hydroperoxide, acetalization with phenol proceeded in poor yield. Attempted acetalizations of 2,3,4,6-tetrabenzylglucose, the corresponding tetraacetate, or their 1-O-silyl derivatives, were unsuccessful (not shown).
Table 2: Transetherification of hemiacetals.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-174-i5.svg?max-width=637&scale=1.0)
|
||||||
| substrate | n | X | R | equiv | product | yielda |
|---|---|---|---|---|---|---|
| 3 | 1 | OH | Ph(CH2)2 | 2 | 5 | 83% |
| 3 | 1 | OH | t-BuO (t-BuOOH) | 2 | 6 | 68–77% |
| 4a | 2 | OH | Ph(CH2)2 | 5 | 7 | 89% |
| 4b | 2 | OTBS | Ph(CH2)2 | 2 | 7 | 82% |
| 4a | 2 | OH | t-BuO | 2 | 8 | 71–80% |
| 4a | 2 | OH | 2-octyl | 2 | 9 | 70–90% |
| 4a | 2 | OH | Ph | 2 | 10 | 7% |
| 4b | 2 | OTBS | Ph | 1 | 10 | 44%b |
aDuplicate values; range given in case of variance. bIncludes some inseparable impurities.
Resubmission of purified acetal 7 to typical reaction conditions in the presence of a slight excess of water did not result in the reformation of 4a. However, we did observe rapid transacetalization when 7 was resubmitted to reaction conditions in the presence of methanol (Scheme 1).
![[1860-5397-9-174-i1]](/bjoc/content/inline/1860-5397-9-174-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Transacetalization of acetal 7.
Scheme 1: Transacetalization of acetal 7.
After TLC monitoring of reactions revealed the build-up of an unknown intermediate, we conducted control reactions in the absence of a nucleophile. These revealed a rapid Re2O7-promoted condensation of the hemiacetals to dimeric oxybisacetals (Table 3). As will be described later, these apparent byproducts proved to be competent substrates for acetal formation.
Table 3: Dimerization of hemiacetals.
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-174-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | X | n | reaction time (h) | conditionsa | bisacetal (%) | aldehyde (%) |
|---|---|---|---|---|---|---|
| 1 | 4a | 2 | 0.1 | a | 11 (57%) | 12 (5%) |
| 2 | 4a | 2 | 0.1 | b | 11 (59%) | 12 (3%) |
| 3 | 4a | 2 | 0.5 | a,d | 11 (12%) | trace |
| 4 | 4a | 2 | 2 | a,e | NR | NR |
| 5 | 3 | 1 | 0.1 | a | 13 (37%) | 14 (28%) |
| 6 | 4a | 2 | 0.1 | a | 11 (30%) | 12 (2%) |
| 7 | 4a | 2 | 0.5 | c | 11 (62%) | 12 (4%) |
aa:1% Re2O7; b: 2% Re2O7; c:10% PTSA; d: with 3 Å mol sieves (powdered); e: with 4 Å mol sieves (powdered).
We next attempted to maximize the yield of acetal based upon alcohol (Table 4). Good yields were obtained at a 1:1 ratio of alcohol to hemiacetal and yields did not vary significantly with the rate of addition of hemiacetal. This latter observation was initially surprising given the rapidity of hemiacetal dimerization (vide supra). However, we soon realized that the bisacetal ether 11 (Table 4, entry 4) was a remarkably effective substrate. In fact, the reaction of phenethyl alcohol (1.0 equiv) with only 0.5 equivalent of the oxybisacetal dimer proceeded rapidly to furnish high yields of 7 (not shown).
Table 4: Optimization of acetalization conditions.
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-174-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | substrate | rate of 4a addition (equiv/min) | reaction time (h) | additives | 7 (%) |
|---|---|---|---|---|---|
| 1 | 4a | 0.1 | 1 | — | 93 |
| 2 | 4a | 0.04 | 1 | — | 94 |
| 3 | 4a | 0.01 | 2 | — | 87 |
| 4 | 11 | all at once | 0.5 | — | 87 |
| 5 | 4a | all at once | 1 | — | 89 |
| 6 | 4a | 0.02 | 1 | 3 Å mol sieves (powder) | NR |
| 7 | 4a | 0.02 | 2 | 4 Å mol sieves (powder) | NR |
Monothioacetal formation
We next turned our attention to the synthesis of monothioacetals, a functionality important for carbonyl protection and as a precursor for oxycarbenium ions [21,22]. As illustrated in Table 5, tetrahydrofuranol and tetrahydropyranol both undergo condensation with a simple thiol in the presence of Re2O7 to provide the monothioacetal in good yield; only traces of the S,S-dithioacetal were observed. The same reaction, when catalyzed by a Brønsted acid, required a higher catalyst loading and provided a lower yield of product accompanied by a greater amount of dithioacetal. Re(VII)-promoted reaction with thioacetic acid proceeded in much lower yield.
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-174-i8.svg?max-width=637&scale=1.0)
As illustrated in Scheme 2, Re2O7 also promotes the reaction of an aldehyde with a thiol or a dithiol to furnish a dithioacetal or a 1,3-dithiane. A ketone substrate did not react under these conditions (not shown).
![[1860-5397-9-174-i2]](/bjoc/content/inline/1860-5397-9-174-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Thioacetalization of hexanal with Re2O7.
Scheme 2: Thioacetalization of hexanal with Re2O7.
The mono- and dithioacetalization reactions were visibly different from the O-acetalizations described above. Whereas addition of Re2O7 to a hemiacetal in the presence of an alcohol generates a transparent light yellow solution (Figure S1, Supporting Information File 1), addition of thiol to a mixture of Re2O7 and hemiacetal produces an opaque, black, solution that gradually becomes translucent but remains very dark (Figure S2, Supporting Information File 1).
Attempted reaction with nitrogen nucleophiles
Perrhenate proved ineffective for catalyzing the formation of N,O-acetals (Table 6). Although these investigations were complicated by the limited solubility of some of the nucleophiles in dichloromethane, similar results were obtained in acetonitrile, where solubility was less of an issue.
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-174-i9.svg?max-width=637&scale=1.0)
The poor results with N-nucleophiles led us to reinvestigate a proven reaction in the presence of amines. The previously successful condensation of 4a with phenethyl alcohol (see Table 2) failed completely in the presence of added morpholine or pyridine (not shown).
Allylation of hemiacetals
Re2O7 has been successfully applied to intramolecular Prins reactions of alkenes with reversibly generated hemiacetals [13], and we were curious about the potential for applications to intermolecular allylations. As illustrated in Table 7, tetrahydrofuranol 3, tetrahydropyranol 4a, bisacetal 11, and oxadadamantyl hemiacetal 22 all underwent allylation in the presence of stoichiometric allyltrimethylsilane and 1% of Re2O7. The isolated yields of 23 and 24 may be artificially low due to product volatility. Peroxyhemiacetal (dioxolanol 1a) failed to react under these conditions.
Table 7: Allylation of hemiacetals.
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-174-i10.svg?max-width=637&scale=1.0)
|
||
| substrate | product | yield |
|---|---|---|
| 1a | NR | NR |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-174-i11.svg?max-width=637&scale=1.0)
23a |
33%b |
| 4a |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-174-i12.svg?max-width=637&scale=1.0)
24a |
41%b |
| 11 | 24a | 53%c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-174-i13.svg?max-width=637&scale=1.0)
22 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-174-i14.svg?max-width=637&scale=1.0)
25 |
90%c |
aVolatile product. bIsolated yield. cGC yield (toluene standard).
Transetherification of hydroperoxyacetals
We were curious about the interactions of Re(VII) oxides with hydroperoxyacetals (1-alkoxyhydroperoxides), versatile intermediates available from ozonolysis of alkenes in alcoholic solvents [23,24]. As can be seen in Table 8, Re2O7, Me3SiOReO3 and PTSA all catalyze alkoxide metathesis to furnish a moderate yield of a new hydroperoxyacetal. The exchange reaction was observed in several solvents (e.g., CH2Cl2, 1,2-dichloroethane) but was most efficient in acetonitrile. The alkoxide exchange was accompanied by much slower exchange of the peroxide; for example, prolonged reaction (>24 h) of acetal 26 in MeOH (solvent) in the presence of catalytic Re(VII) furnished a 40% yield of the dimethyl acetal (not shown).
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-174-i15.svg?max-width=637&scale=1.0)
Discussion
All the reactions described appear to involve the intermediacy of perrhenate esters. Previous investigators have hypothesized that the barrier for Re2O7-promoted C–O ionization of hemiacetals is relatively low [13]. The differences in reactivity between hemiacetals of cyclic ethers (displaced by alcohols and allyltrimethylsilane) and those of cyclic peroxides (reactive only towards alcohols) demonstrates that the extent of activation is dependent on the nature of the substrate, and that different levels of activation are required for trapping by heteroatom versus carbon nucleophiles. Previous work established that the perrhenate-catalyzed isomerization of alcohols can also employ silyl ethers as substrates [25], and our work demonstrates that the same is true for the hemiacetals investigated here. However, our work also demonstrates that oxybisacetals (for example, bis 2-tetrahydropyranyl ether) are highly effective substrates for the Re-catalyzed processes.
The results are in keeping with the formation of perrhenate esters which can undergo displacement by nucleophiles. Our results suggest that in the case of etherifications with alcohols, the intermediates can sometimes be regenerated from the product acetals. Moreover, our results clearly demonstrate that Re2O7 reversibly dimerizes hemiacetals in a reaction that is sufficiently rapid that it is possible that the oxybisacetal dimers may be the predominant precursors of the perrhenate intermediates and therefore the reaction products (Scheme 3).
![[1860-5397-9-174-i3]](/bjoc/content/inline/1860-5397-9-174-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The results with peroxyhemiacetals are consistent with the known reactivity of bisperoxyacetals [26]. The hydroperoxyacetals showed no sign of electrophilic activation of the hydroperoxide (which would presumably lead to heterolytic fragmentation) and activation of the peroxide C–O was clearly disfavored relative to activation of the alkoxide C–O bond. Rapid alkoxide metathesis was also observed in the presence of a strong Brønsted acid. Our observations suggest that the seeming lack of reactivity of ozonolysis-derived 1-methoxyhydroperoxides towards methanolic acid may mask a rapid degenerate exchange [27].
The failure of the perrhenate to catalyze formation of N,O-acetals was initially perplexing. However, reported perrhenate-promoted dehydrations of amides and oximes require relatively high temperatures [28-30]. Our results suggest that amines and amides actively suppress the ability of Re(VII)-oxides to activate alcohols. We note, however, a recent report by Ghorai describing the allylation of iminium ions generated from aldehydes and sulfonamides in the presence of Re2O7 [15].
Conclusion
Perrhenates hold broad potential as catalysts for electrophilic activation of hemiacetals. The ability to catalyze formation of O,O-, O,S-, and S,S-acetals under very mild conditions offers a useful complement to traditional Brønsted acid catalysts.
Supporting Information
Experimental details and detailed information, including references and spectral listings related to prepared molecules, are provided in Supporting Information File 1.
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 562.7 KB | Download |
References
-
Chemla, F.; Ferreira, F.; Rey, B. Acetals: Hal/X and O/O, S, Se, Te. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Ley, S., Ed.; George Thieme Verlag: Stuttgart, 2007; Vol. 29, pp 801–887.
Return to citation in text: [1] -
Mol, J. C. Catal. Today 1999, 51, 289–299. doi:10.1016/S0920-5861(99)00051-6
Return to citation in text: [1] -
Bellemin-Laponnaz, S. ChemCatChem 2009, 1, 357–362. doi:10.1002/cctc.200900206
Return to citation in text: [1] -
Dussault, P. H.; Ghorai, P. Rhenium(VII) Oxide. In e-EROS Encylopedia of Reagents for Organic Synthesis, Wiley, 2010. doi:10.1002/047084289X
Return to citation in text: [1] -
Volchkov, I.; Lee, D. J. Am. Chem. Soc. 2013, 135, 5324–5327. doi:10.1021/ja401717b
Return to citation in text: [1] -
Xie, Y.; Floreancig, P. E. Angew. Chem., Int. Ed. 2013, 52, 625–628. doi:10.1002/anie.201208132
Return to citation in text: [1] -
Volchkov, I.; Park, S.; Lee, D. Org. Lett. 2011, 13, 3530–3533. doi:10.1021/ol2013473
Return to citation in text: [1] -
Xie, Y.; Floreancig, P. E. Chem. Sci. 2011, 2, 2423–2427. doi:10.1039/c1sc00570g
Return to citation in text: [1] [2] -
Stivala, C. E.; Gu, Z.; Smith, L. L.; Zakarian, A. Org. Lett. 2012, 14, 804–807. doi:10.1021/ol203342e
Return to citation in text: [1] -
Herrmann, A. T.; Saito, T.; Stivala, C. E.; Tom, J.; Zakarian, A. J. Am. Chem. Soc. 2010, 132, 5962–5963. doi:10.1021/ja101673v
Return to citation in text: [1] -
Ghorai, P.; Dussault, P. H. Org. Lett. 2009, 11, 213–216. doi:10.1021/ol8023874
Return to citation in text: [1] -
Ghorai, P.; Dussault, P. H. Org. Lett. 2008, 10, 4577–4579. doi:10.1021/ol801859c
Return to citation in text: [1] -
Tadpetch, K.; Rychnosvky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204
Return to citation in text: [1] [2] [3] -
Das, B. G.; Nallagonda, R.; Ghorai, P. J. Org. Chem. 2012, 77, 5577–5583. doi:10.1021/jo300706b
Return to citation in text: [1] -
Pramanik, S.; Ghorai, P. Chem. Commun. 2012, 48, 1820–1822. doi:10.1039/c2cc15472b
Return to citation in text: [1] [2] -
Schiaffo, C. E.; Rottman, M.; Wittlin, S.; Dussault, P. H. ACS Med. Chem. Lett. 2011, 2, 316–319. doi:10.1021/ml100308d
Return to citation in text: [1] -
Ingram, K.; Schiaffo, C. E.; Sittiwong, W.; Benner, E.; Dussault, P. H.; Keiser, J. J. Antimicrob. Chemother. 2012, 67, 1979–1986. doi:10.1093/jac/dks141
Return to citation in text: [1] -
Dussault, P. H.; Liu, X. Org. Lett. 1999, 1, 1391–1393. doi:10.1021/ol990954y
Return to citation in text: [1] -
Bellemin-Laponnaz, S.; Gisie, H.; Le Ny, J. P.; Osborn, J. A. Angew. Chem., Int. Ed. Engl. 1997, 36, 976–978. doi:10.1002/anie.199709761
Return to citation in text: [1] -
Edwards, P.; Wilkinson, G. J. Chem. Soc., Dalton Trans. 1984, 2695–2702. doi:10.1039/DT9840002695
Return to citation in text: [1] -
Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons, Inc.: Hoboken, 2007.
Return to citation in text: [1] -
Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015–9020. doi:10.1021/ja0111481
Return to citation in text: [1] -
Bunnelle, W. H. Chem. Rev. 1991, 91, 335–362. doi:10.1021/cr00003a003
Return to citation in text: [1] -
Yamamoto, Y.; Niki, E.; Kamiya, Y. Bull. Chem. Soc. Jpn. 1982, 55, 2677–2678. doi:10.1246/bcsj.55.2677
Return to citation in text: [1] -
Bellemin-Laponnaz, S.; Le Ny, J. P.; Osborn, J. A. Tetrahedron Lett. 2000, 41, 1549–1552. doi:10.1016/S0040-4039(99)02352-7
Return to citation in text: [1] -
Dussault, P. H.; Lee, I. Q.; Lee, H.-J.; Lee, R. J.; Niu, Q. J.; Schultz, J. A.; Zope, U. R. J. Org. Chem. 2000, 65, 8407–8414. doi:10.1021/jo991714z
Return to citation in text: [1] -
Claus, R. E.; Schreiber, S. L. Org. Synth. 1986, 64, 150–156.
Return to citation in text: [1] -
Furuya, Y.; Ishihara, K.; Yamamoto, H. Bull. Chem. Soc. Jpn. 2007, 80, 400–406. doi:10.1246/bcsj.80.400
Return to citation in text: [1] -
Kusama, H.; Yamashita, Y.; Narasaka, K. Bull. Chem. Soc. Jpn. 1995, 68, 373–377. doi:10.1246/bcsj.68.373
Return to citation in text: [1] -
Kusama, H.; Yamashita, Y.; Uchiyama, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1997, 70, 965–975. doi:10.1246/bcsj.70.965
Return to citation in text: [1]
| 15. | Pramanik, S.; Ghorai, P. Chem. Commun. 2012, 48, 1820–1822. doi:10.1039/c2cc15472b |
| 28. | Furuya, Y.; Ishihara, K.; Yamamoto, H. Bull. Chem. Soc. Jpn. 2007, 80, 400–406. doi:10.1246/bcsj.80.400 |
| 29. | Kusama, H.; Yamashita, Y.; Narasaka, K. Bull. Chem. Soc. Jpn. 1995, 68, 373–377. doi:10.1246/bcsj.68.373 |
| 30. | Kusama, H.; Yamashita, Y.; Uchiyama, K.; Narasaka, K. Bull. Chem. Soc. Jpn. 1997, 70, 965–975. doi:10.1246/bcsj.70.965 |
| 1. | Chemla, F.; Ferreira, F.; Rey, B. Acetals: Hal/X and O/O, S, Se, Te. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Ley, S., Ed.; George Thieme Verlag: Stuttgart, 2007; Vol. 29, pp 801–887. |
| 8. | Xie, Y.; Floreancig, P. E. Chem. Sci. 2011, 2, 2423–2427. doi:10.1039/c1sc00570g |
| 13. | Tadpetch, K.; Rychnosvky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204 |
| 25. | Bellemin-Laponnaz, S.; Le Ny, J. P.; Osborn, J. A. Tetrahedron Lett. 2000, 41, 1549–1552. doi:10.1016/S0040-4039(99)02352-7 |
| 11. | Ghorai, P.; Dussault, P. H. Org. Lett. 2009, 11, 213–216. doi:10.1021/ol8023874 |
| 12. | Ghorai, P.; Dussault, P. H. Org. Lett. 2008, 10, 4577–4579. doi:10.1021/ol801859c |
| 26. | Dussault, P. H.; Lee, I. Q.; Lee, H.-J.; Lee, R. J.; Niu, Q. J.; Schultz, J. A.; Zope, U. R. J. Org. Chem. 2000, 65, 8407–8414. doi:10.1021/jo991714z |
| 3. | Bellemin-Laponnaz, S. ChemCatChem 2009, 1, 357–362. doi:10.1002/cctc.200900206 |
| 4. | Dussault, P. H.; Ghorai, P. Rhenium(VII) Oxide. In e-EROS Encylopedia of Reagents for Organic Synthesis, Wiley, 2010. doi:10.1002/047084289X |
| 5. | Volchkov, I.; Lee, D. J. Am. Chem. Soc. 2013, 135, 5324–5327. doi:10.1021/ja401717b |
| 6. | Xie, Y.; Floreancig, P. E. Angew. Chem., Int. Ed. 2013, 52, 625–628. doi:10.1002/anie.201208132 |
| 7. | Volchkov, I.; Park, S.; Lee, D. Org. Lett. 2011, 13, 3530–3533. doi:10.1021/ol2013473 |
| 8. | Xie, Y.; Floreancig, P. E. Chem. Sci. 2011, 2, 2423–2427. doi:10.1039/c1sc00570g |
| 9. | Stivala, C. E.; Gu, Z.; Smith, L. L.; Zakarian, A. Org. Lett. 2012, 14, 804–807. doi:10.1021/ol203342e |
| 10. | Herrmann, A. T.; Saito, T.; Stivala, C. E.; Tom, J.; Zakarian, A. J. Am. Chem. Soc. 2010, 132, 5962–5963. doi:10.1021/ja101673v |
| 23. | Bunnelle, W. H. Chem. Rev. 1991, 91, 335–362. doi:10.1021/cr00003a003 |
| 24. | Yamamoto, Y.; Niki, E.; Kamiya, Y. Bull. Chem. Soc. Jpn. 1982, 55, 2677–2678. doi:10.1246/bcsj.55.2677 |
| 13. | Tadpetch, K.; Rychnosvky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204 |
| 18. | Dussault, P. H.; Liu, X. Org. Lett. 1999, 1, 1391–1393. doi:10.1021/ol990954y |
| 21. | Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons, Inc.: Hoboken, 2007. |
| 22. | Crich, D.; Smith, M. J. Am. Chem. Soc. 2001, 123, 9015–9020. doi:10.1021/ja0111481 |
| 16. | Schiaffo, C. E.; Rottman, M.; Wittlin, S.; Dussault, P. H. ACS Med. Chem. Lett. 2011, 2, 316–319. doi:10.1021/ml100308d |
| 17. | Ingram, K.; Schiaffo, C. E.; Sittiwong, W.; Benner, E.; Dussault, P. H.; Keiser, J. J. Antimicrob. Chemother. 2012, 67, 1979–1986. doi:10.1093/jac/dks141 |
| 13. | Tadpetch, K.; Rychnosvky, S. D. Org. Lett. 2008, 10, 4839–4842. doi:10.1021/ol8019204 |
| 15. | Pramanik, S.; Ghorai, P. Chem. Commun. 2012, 48, 1820–1822. doi:10.1039/c2cc15472b |
| 14. | Das, B. G.; Nallagonda, R.; Ghorai, P. J. Org. Chem. 2012, 77, 5577–5583. doi:10.1021/jo300706b |
| 19. | Bellemin-Laponnaz, S.; Gisie, H.; Le Ny, J. P.; Osborn, J. A. Angew. Chem., Int. Ed. Engl. 1997, 36, 976–978. doi:10.1002/anie.199709761 |
| 20. | Edwards, P.; Wilkinson, G. J. Chem. Soc., Dalton Trans. 1984, 2695–2702. doi:10.1039/DT9840002695 |
© 2013 Sittiwong et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)