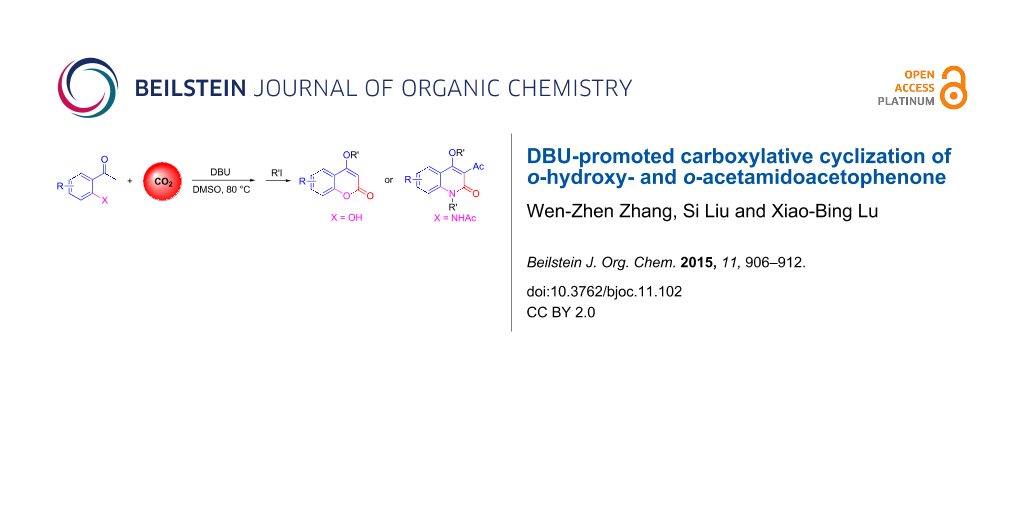
Abstract
The carboxylative cyclization of o-hydroxy- and o-acetamidoacetophenone with carbon dioxide promoted by the organic base 1,8-diazabicycloundec-7-ene (DBU) is reported. This reaction provides convenient access to the biologically important compounds 4-hydroxy-2H-chromen-2-one and 4-hydroxy-2(1H)-quinolinone in moderate to good yields using carbon dioxide as the carboxylation reagent. An acyl migration from nitrogen to carbon is observed in the reaction of o-acetamidoacetophenone.

Graphical Abstract
Introduction
4-Hydroxy-2H-chromen-2-ones and 4-hydroxy-2(1H)-quinolinones are key structural subunits found in many natural products [1], commercial drugs [2,3] and pharmacologically potent compounds (Figure 1) [4,5]. Warfarin, for example, is an anticoagulant widely used to prevent thrombosis [2]; Novobiocin has long been established as an aminocoumarin antibiotic [3]. Recent studies revealed that the anticoagulant Dicumarol is able to inhibit the growth of pancreatic cancer [4]. Roquinimex was reported as an antineoplastic agent [5]. Traditional methods for accessing these compounds rely heavily on cyclization reactions using diethyl carbonate in the presence of inorganic bases [6,7] or Friedel–Crafts reactions using strong and corrosive acids [8]. In terms of availability and toxicity of the starting materials, environmental benignity and economical concerns, the development of an alternative method for the synthesis of these compounds using carbon dioxide as the carboxylation reagent [9-16] is highly desirable.
![[1860-5397-11-102-1]](/bjoc/content/figures/1860-5397-11-102-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples for biologically active 4-hydroxy-2H-chromen-2-one and 4-hydroxy-2(1H)-quinolinone compounds.
Figure 1: Selected examples for biologically active 4-hydroxy-2H-chromen-2-one and 4-hydroxy-2(1H)-quinolinon...
It was previously reported that the α C–H bond in aromatic ketones readily undergoes a carboxylation reaction with carbon dioxide in the presence of a suitable base, producing β-ketocarboxylic acids [17-20]. Given that o-hydroxy- or o-acetamidoacetophenone is used as the starting material to react with carbon dioxide, the intramolecular carboxylative cyclization might provide a convenient access to 4-hydroxy-2H-chromen-2-one and 4-hydroxy-2(1H)-quinolinone. Indeed, Da Re and Sandri reported in 1960 that o-hydroxyacetophenone derivatives react with carbon dioxide (4 MPa) in the presence of 3 equivalents of potassium carbonate at 130–170 °C, yielding 4-hydroxy-2H-chromen-2-ones in moderate yields [21]. From the viewpoints of solubility, efficiency, and ease of recovery and reuse, the use of an organic base rather than potassium carbonate in this reaction would be more promising. DBU and MTBD were previously reported as suitable bases to promote the carboxylation of α-C–H bonds in aromatic ketones with carbon dioxide [17-20]. In extension of our continuous efforts in developing catalytic transformations of carbon dioxide into value-added fine chemicals [20,22,23], we report herein the DBU-promoted carboxylative cyclization of o-hydroxy- and o-acetamidoacetophenones with carbon dioxide to give 4-hydroxy-2H-chromen-2-ones and 4-hydroxy-2(1H)-quinolinones, respectively, in moderate to good yields under mild reaction conditions. An acyl migration from the nitrogen to carbon is observed in the reaction of o-acetamidoacetophenone.
Results and Discussion
We started our investigation with the carboxylative cyclization of o-hydroxypropiophenone (1a) with carbon dioxide to identify the optimal organic base and reaction conditions (Table 1). The use of potassium carbonate as base in DMF at 100 °C gave 29% yield of product 2a (Table 1, entry 1). When DBU and MTBD were used in this reaction instead of potassium carbonate, a significantly increased yield of 2a was obtained (Table 1, entries 2 and 3). When switching the solvent to DMSO, further increased yields were obtained, whereby DBU showed a higher efficiency than MTBD (Table 1, entries 4 and 5). Other solvents such as DMAc and THF gave dramatically decreased yields (Table 1, entries 6 and 7). Unexpectedly, we found that a decrease of temperature from 100 °C to 80 °C in DMSO led to a higher yield (87%) of 2a (Table 1, entry 8). The reaction was found to be sensitive to the carbon dioxide pressure and performing the reaction at a lower pressure gave a distinctly decreased yield (Table 1, entry 10). When the reaction was conducted under atmospheric carbon dioxide, no carboxylative cyclization product was obtained (Table 1, entry 11). Therefore, the optimal reaction conditions were established as following: 2.0 equiv DBU as base, 3.0 MPa of carbon dioxide, DMSO as solvent at 80 °C for 24 h.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-102-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvent | T/°C | p(CO2)/MPa | Yield/%b |
|---|---|---|---|---|---|
| 1 | K2CO3 | DMF | 100 | 3 | 29 |
| 2 | DBU | DMF | 100 | 3 | 49 |
| 3 | MTBD | DMF | 100 | 3 | 65 |
| 4 | MTBD | DMSO | 100 | 3 | 68 |
| 5 | DBU | DMSO | 100 | 3 | 75 |
| 6 | DBU | DMAc | 100 | 3 | 32 |
| 7 | DBU | THF | 100 | 3 | 10 |
| 8 | DBU | DMSO | 80 | 3 | 87 |
| 9 | DBU | DMSO | 60 | 3 | 65 |
| 10 | DBU | DMSO | 80 | 2 | 53 |
| 11 | DBU | DMSO | 80 | 0.1 | <1 |
aReaction conditions: o-hydroxyacetophenone (1a, 0.5 mmol), base (1 mmol), solvent (2 mL), 24 h; then n-BuI (1.0 mmol), 80 °C, 4 h. bIsolated yield.
Under the optimal reaction conditions, the substrate scope was then investigated (Table 2). Compared with o-hydroxypropiophenone, o-hydroxyacetophenone gave a slightly lower yield of the 2H-chromen-2-one product (Table 2, entries 2 and 4). o-Hydroxyacetophenone bearing electron-donating alkyl and ether groups, or electron-withdrawing fluoro and bromo groups undergoes the carboxylative cyclization reaction smoothly, affording the corresponding 4-butoxy-2H-chromen-2-ones 2b–2f in moderate to good yields (Table 2, entries 2–6). The bromo group in product 2f and the alkyne group in product 2g offer opportunities for further functionalization of these 2H-chromen-2-ones using well-established methods [24] (Table 2, entries 6 and 7). 2-Hydroxy-1-acetylnaphthalene (1h) participates in the carboxylative cyclization reaction to furnish the tricyclic product 2h in moderate yield (Table 2, entry 8).
Table 2: Carboxylative cyclization of various o-hydroxyacetophenones with carbon dioxide.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-102-i4.svg?max-width=637&scale=1.0)
|
|||
| Enty | Substrate | Product | Yield/%b |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-102-i5.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-102-i6.svg?max-width=637&scale=1.0)
2a |
87 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-102-i7.svg?max-width=637&scale=1.0)
1b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-102-i8.svg?max-width=637&scale=1.0)
2b |
79 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-102-i9.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-102-i10.svg?max-width=637&scale=1.0)
2c |
56 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-102-i11.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-102-i12.svg?max-width=637&scale=1.0)
2d |
45 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-102-i13.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-102-i14.svg?max-width=637&scale=1.0)
2e |
49 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-102-i15.svg?max-width=637&scale=1.0)
1f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-102-i16.svg?max-width=637&scale=1.0)
2f |
36 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-102-i17.svg?max-width=637&scale=1.0)
1g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-102-i18.svg?max-width=637&scale=1.0)
2g |
65 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-102-i19.svg?max-width=637&scale=1.0)
1h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-102-i20.svg?max-width=637&scale=1.0)
2h |
42 |
aReaction conditions: o-hydroxyacetophenone (1) (0.5 mmol), DBU (1.0 mmol), CO2 (3.0 MPa), DMSO (2 mL), 80 °C, 24 h; then n-BuI (1.0 mmol), 80 °C, 4 h. bIsolated yield.
With the successful DBU-promoted carboxylative cyclization of o-hydroxyacetophenone at hand, we then extended this strategy to o-acetamidoacetophenone to synthesize 4-hydroxy-2(1H)-quinolinone (Table 3). Using 4 equivalents DBU as base in DMSO at 80 °C, o-acetamidoacetophenone (3a) underwent the carboxylative cyclization reaction to provide 3-acetyl-4-methoxy-2(1H)-quinolinones 4a and 5a (Table 3, entry 1). Noteworthy, the acyl group was no longer bound to nitrogen in the product, which implies that a nitrogen to carbon acyl migration occurred during the reaction. The derivatization reaction using iodide compounds at higher temperature led to complex product mixtures. o-Acetamidoacetophenone substrates containing methoxy (3b) and bromo (3c) groups also reacted smoothly to afford the corresponding products (Table 3, entries 2 and 3). The reactions using benzamido- (3d) and p-toluenesulfonamido- (3e) acetophenone gave complex mixtures and no carboxylative cyclization product was observed (Table 3, entries 4 and 5).
Table 3: Carboxylative cyclization of various o-acetamidoacetophenones with carbon dioxide.a
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-102-i21.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | Product | Yield/% | |
|---|---|---|---|---|
| 4 | 5 | 4 + 5 | ||
| 1 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-102-i22.svg?max-width=637&scale=1.0)
3a |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-102-i23.svg?max-width=637&scale=1.0)
4a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-102-i24.svg?max-width=637&scale=1.0)
5a |
42 + 35 |
| 2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-102-i25.svg?max-width=637&scale=1.0)
3b |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-102-i26.svg?max-width=637&scale=1.0)
4b |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-102-i27.svg?max-width=637&scale=1.0)
5b |
38 + 37 |
| 3 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-102-i28.svg?max-width=637&scale=1.0)
3c |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-102-i29.svg?max-width=637&scale=1.0)
4c |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-102-i30.svg?max-width=637&scale=1.0)
5c |
32 + 20 |
| 4 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-102-i31.svg?max-width=637&scale=1.0)
3d |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-102-i32.svg?max-width=637&scale=1.0)
4d |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-102-i33.svg?max-width=637&scale=1.0)
5d |
<1 |
| 5 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-102-i34.svg?max-width=637&scale=1.0)
3e |
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-102-i35.svg?max-width=637&scale=1.0)
4e |
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-102-i36.svg?max-width=637&scale=1.0)
5e |
<1 |
aReaction conditions: o-acetamidoacetophenone (3, 0.5 mmol), DBU (2.0 mmol), CO2 (3.0 MPa), DMSO (2 mL), 80 °C, 24 h; then MeI (2.0 mmol), 30 °C, 4 h. bIsolated yield of separated products.
A likely mechanism for the carboxylative cyclization of o-acetamidoacetophenone with carbon dioxide is proposed as shown in Scheme 1. The reaction can evolve along two pathways: in path A, deprotonation of o-acetamidoacetophenone by DBU gives enolate I, which undergoes an acyl migration from nitrogen to carbon [25,26] similar to the Baker–Venkataraman O to C acyl migration [27]. After a proton shift from the enol to nitrogen, the resultant intermediate III is carboxylated with carbon dioxide in the presence of DBU to afford intermediate IV, which subsequently undergoes a cyclization reaction to give V. The product is obtained after derivatization with methyl iodide. Also, path B in which the N to C acyl migration occurs after the carboxylative cyclization cannot be excluded.
![[1860-5397-11-102-i1]](/bjoc/content/inline/1860-5397-11-102-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Possible mechanism for the carboxylative cyclization of o-acetamidoacetophenone.
Scheme 1: Possible mechanism for the carboxylative cyclization of o-acetamidoacetophenone.
We also conducted a cross experiment as shown in Scheme 2. When compounds 3b and 3f were reacted concomitantly, the corresponding carboxylative cyclization products 4b and 4f were obtained. No cross products 6 and 7 were detected, which implies that the N to C acyl shift occurred intramolecularly, not intermolecularly.
![[1860-5397-11-102-i2]](/bjoc/content/inline/1860-5397-11-102-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cross carboxylative cyclization reaction.
Scheme 2: Cross carboxylative cyclization reaction.
Conclusion
In summary, we have developed a DBU-promoted carboxylative cyclization of o-hydroxy- and o-acetamidoacetophenones with carbon dioxide. This methodology provides a convenient access to the biologically important 4-hydroxy-2H-chromen-2-ones and 4-hydroxy-2(1H)-quinolinones in moderate to good yields under mild reaction conditions. While there are precedents for the carboxylation of enolates, a practical protocol was developed that relies on in situ cyclization to form thermodynamically stable coumarins. Importantly, the use of an intramolecular in situ trap avoids the problem of decarboxylation during workup. In case of o-acetamidoacetophenones, an acyl migration from nitrogen to carbon was observed. The cross experiment showed that the N to C acyl shift occurred intramolecularly.
Experimental
Similarly as described in our previous paper [22], a 20 mL oven-dried autoclave containing a stirring bar was charged with o-hydroxyacetophenone (1) or o-acetamidoacetophenone (3) (0.5 mmol), DBU (1.0 mmol for 1, 2.0 mmol for 3), and 2 mL dry DMSO. After purging the autoclave with CO2 three times, the sealed autoclave was pressurized to the appropriate pressure with CO2. The reaction mixture was stirred at 80 °C for 24 h, then the autoclave was cooled to room temperature and the remaining CO2 was vented slowly. Then n-BuI (1.0 mmol for 1) or MeI (2.0 mmol for 3) was added into the autoclave and the reaction mixture was stirred at 80 °C (for 1) or at 30 °C (for 3) for 4 h. The reaction mixture was then diluted with water (30 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with water and brine, dried over Na2SO4 and filtered. The solvent was removed under vacuum. The product was isolated by column chromatography on silica gel (hexane/ethyl acetate 2:1).
Supporting Information
| Supporting Information File 1: Experimental procedures, spectroscopic and analytical data, and copies of NMR spectra of the products. | ||
| Format: PDF | Size: 802.5 KB | Download |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21172026), the Fundamental Research Funds for the Central Universities (DUT15LAB21), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13008). X.-B. Lu gratefully acknowledges the Chang Jiang Scholars Program (no. T2011056) from Ministry of Education, People’s Republic of China.
References
-
Pratap, R.; Ram, V. J. Chem. Rev. 2014, 114, 10476. doi:10.1021/cr500075s
See for a review.
Return to citation in text: [1] -
Holbrook, A. M.; Pereira, J. A.; Labiris, R.; McDonald, H.; Douketis, J. D.; Crowther, M.; Wells, P. S. Arch. Intern. Med. 2005, 165, 1095. doi:10.1001/archinte.165.10.1095
Return to citation in text: [1] [2] -
Hoeksema, H.; Johnson, J. L.; Hinman, J. W. J. Am. Chem. Soc. 1955, 77, 6710. doi:10.1021/ja01629a129
Return to citation in text: [1] [2] -
Cullen, J. J.; Hinkhouse, M. M.; Grady, M.; Gaut, A. W.; Liu, J.; Zhang, Y. P.; Weydert, C. J. D.; Domann, F. E.; Oberley, L. W. Cancer Res. 2003, 63, 5513.
Return to citation in text: [1] [2] -
Jönsson, S.; Andersson, G.; Fex, T.; Fristedt, T.; Hedlund, G.; Jansson, K.; Abramo, L.; Fritzson, I.; Pekarski, O.; Runström, A.; Sandin, H.; Thuvesson, I.; Björk, A. J. Med. Chem. 2004, 47, 2075. doi:10.1021/jm031044w
Return to citation in text: [1] [2] -
Hack, D.; Chauhan, P.; Deckers, K.; Hermann, G. N.; Mertens, L.; Raabe, G.; Enders, D. Org. Lett. 2014, 16, 5188. doi:10.1021/ol502551u
Return to citation in text: [1] -
Prousis, K. C.; Tzani, A.; Avlonitis, N.; Calogeropoulou, T.; Detsi, A. J. Heterocycl. Chem. 2013, 50, 1313. doi:10.1002/jhet.1869
Return to citation in text: [1] -
Ambre, P. K.; Pissurlenkar, R. R. S.; Wavhale, R. D.; Shaikh, M. S.; Khedkar, V. M.; Wan, B.; Franzblau, S. G.; Coutinho, E. C. Med. Chem. Res. 2014, 23, 2564. doi:10.1007/s00044-013-0850-7
Return to citation in text: [1] -
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi:10.1021/cr068357u
Return to citation in text: [1] -
Riduan, S. N.; Zhang, Y. Dalton Trans. 2010, 39, 3347. doi:10.1039/b920163g
Return to citation in text: [1] -
Huang, K.; Sun, C.-L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435. doi:10.1039/c0cs00129e
Return to citation in text: [1] -
Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W. A.; Kühn, F. E. Angew. Chem., Int. Ed. 2011, 50, 8510. doi:10.1002/anie.201102010
Return to citation in text: [1] -
Zhang, W.; Lü, X. Chin. J. Catal. 2012, 33, 745. doi:10.1016/S1872-2067(11)60390-2
Return to citation in text: [1] -
Tsuji, Y.; Fujihara, T. Chem. Commun. 2012, 48, 9956. doi:10.1039/c2cc33848c
Return to citation in text: [1] -
Zhang, L.; Hou, Z. Chem. Sci. 2013, 4, 3395. doi:10.1039/c3sc51070k
Return to citation in text: [1] -
Aresta, M.; Dibenedetto, A.; Angelini, A. Chem. Rev. 2014, 114, 1709. doi:10.1021/cr4002758
Return to citation in text: [1] -
Flowers, B. J.; Gautreau-Service, R.; Jessop, P. G. Adv. Synth. Catal. 2008, 350, 2947. doi:10.1002/adsc.200800516
Return to citation in text: [1] [2] -
Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. Angew. Chem., Int. Ed. 2012, 51, 6989. doi:10.1002/anie.201201399
Return to citation in text: [1] [2] -
Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Chem. Commun. 2013, 49, 11320. doi:10.1039/c3cc47221c
Return to citation in text: [1] [2] -
Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Org. Chem. Front. 2014, 1, 275. doi:10.1039/c3qo00047h
Return to citation in text: [1] [2] [3] -
Da Re, P.; Sandri, E. Chem. Ber. 1960, 93, 1085. doi:10.1002/cber.19600930514
Return to citation in text: [1] -
Guo, C.-X.; Zhang, W.-Z.; Liu, S.; Lu, X.-B. Catal. Sci. Technol. 2014, 4, 1570. doi:10.1039/c3cy00858d
Return to citation in text: [1] [2] -
Zhang, W.-Z.; Xia, T.; Yang, X.-T.; Lu, X.-B. Chem. Commun. 2015, 51, 6175. doi:10.1039/c5cc01530h
Return to citation in text: [1] -
Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2010, 49, 9047. doi:10.1002/anie.201006374
See for a review.
Return to citation in text: [1] -
Zhang, Z.; Fang, S.; Liu, Q.; Zhang, G. J. Org. Chem. 2012, 77, 7665. doi:10.1021/jo3010217
Return to citation in text: [1] -
Strumfs, B.; Hermane, J.; Belyakov, S.; Trapencieris, P. Tetrahedron 2014, 70, 355. doi:10.1016/j.tet.2013.11.052
Return to citation in text: [1] -
Ameen, D.; Snape, T. J. Synthesis 2015, 47, 141. doi:10.1055/s-0034-1379498
Return to citation in text: [1]
| 22. | Guo, C.-X.; Zhang, W.-Z.; Liu, S.; Lu, X.-B. Catal. Sci. Technol. 2014, 4, 1570. doi:10.1039/c3cy00858d |
| 1. |
Pratap, R.; Ram, V. J. Chem. Rev. 2014, 114, 10476. doi:10.1021/cr500075s
See for a review. |
| 3. | Hoeksema, H.; Johnson, J. L.; Hinman, J. W. J. Am. Chem. Soc. 1955, 77, 6710. doi:10.1021/ja01629a129 |
| 24. |
Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2010, 49, 9047. doi:10.1002/anie.201006374
See for a review. |
| 2. | Holbrook, A. M.; Pereira, J. A.; Labiris, R.; McDonald, H.; Douketis, J. D.; Crowther, M.; Wells, P. S. Arch. Intern. Med. 2005, 165, 1095. doi:10.1001/archinte.165.10.1095 |
| 25. | Zhang, Z.; Fang, S.; Liu, Q.; Zhang, G. J. Org. Chem. 2012, 77, 7665. doi:10.1021/jo3010217 |
| 26. | Strumfs, B.; Hermane, J.; Belyakov, S.; Trapencieris, P. Tetrahedron 2014, 70, 355. doi:10.1016/j.tet.2013.11.052 |
| 4. | Cullen, J. J.; Hinkhouse, M. M.; Grady, M.; Gaut, A. W.; Liu, J.; Zhang, Y. P.; Weydert, C. J. D.; Domann, F. E.; Oberley, L. W. Cancer Res. 2003, 63, 5513. |
| 5. | Jönsson, S.; Andersson, G.; Fex, T.; Fristedt, T.; Hedlund, G.; Jansson, K.; Abramo, L.; Fritzson, I.; Pekarski, O.; Runström, A.; Sandin, H.; Thuvesson, I.; Björk, A. J. Med. Chem. 2004, 47, 2075. doi:10.1021/jm031044w |
| 17. | Flowers, B. J.; Gautreau-Service, R.; Jessop, P. G. Adv. Synth. Catal. 2008, 350, 2947. doi:10.1002/adsc.200800516 |
| 18. | Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. Angew. Chem., Int. Ed. 2012, 51, 6989. doi:10.1002/anie.201201399 |
| 19. | Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Chem. Commun. 2013, 49, 11320. doi:10.1039/c3cc47221c |
| 20. | Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Org. Chem. Front. 2014, 1, 275. doi:10.1039/c3qo00047h |
| 2. | Holbrook, A. M.; Pereira, J. A.; Labiris, R.; McDonald, H.; Douketis, J. D.; Crowther, M.; Wells, P. S. Arch. Intern. Med. 2005, 165, 1095. doi:10.1001/archinte.165.10.1095 |
| 3. | Hoeksema, H.; Johnson, J. L.; Hinman, J. W. J. Am. Chem. Soc. 1955, 77, 6710. doi:10.1021/ja01629a129 |
| 20. | Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Org. Chem. Front. 2014, 1, 275. doi:10.1039/c3qo00047h |
| 22. | Guo, C.-X.; Zhang, W.-Z.; Liu, S.; Lu, X.-B. Catal. Sci. Technol. 2014, 4, 1570. doi:10.1039/c3cy00858d |
| 23. | Zhang, W.-Z.; Xia, T.; Yang, X.-T.; Lu, X.-B. Chem. Commun. 2015, 51, 6175. doi:10.1039/c5cc01530h |
| 8. | Ambre, P. K.; Pissurlenkar, R. R. S.; Wavhale, R. D.; Shaikh, M. S.; Khedkar, V. M.; Wan, B.; Franzblau, S. G.; Coutinho, E. C. Med. Chem. Res. 2014, 23, 2564. doi:10.1007/s00044-013-0850-7 |
| 17. | Flowers, B. J.; Gautreau-Service, R.; Jessop, P. G. Adv. Synth. Catal. 2008, 350, 2947. doi:10.1002/adsc.200800516 |
| 18. | Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. Angew. Chem., Int. Ed. 2012, 51, 6989. doi:10.1002/anie.201201399 |
| 19. | Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Chem. Commun. 2013, 49, 11320. doi:10.1039/c3cc47221c |
| 20. | Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Org. Chem. Front. 2014, 1, 275. doi:10.1039/c3qo00047h |
| 6. | Hack, D.; Chauhan, P.; Deckers, K.; Hermann, G. N.; Mertens, L.; Raabe, G.; Enders, D. Org. Lett. 2014, 16, 5188. doi:10.1021/ol502551u |
| 7. | Prousis, K. C.; Tzani, A.; Avlonitis, N.; Calogeropoulou, T.; Detsi, A. J. Heterocycl. Chem. 2013, 50, 1313. doi:10.1002/jhet.1869 |
| 21. | Da Re, P.; Sandri, E. Chem. Ber. 1960, 93, 1085. doi:10.1002/cber.19600930514 |
| 5. | Jönsson, S.; Andersson, G.; Fex, T.; Fristedt, T.; Hedlund, G.; Jansson, K.; Abramo, L.; Fritzson, I.; Pekarski, O.; Runström, A.; Sandin, H.; Thuvesson, I.; Björk, A. J. Med. Chem. 2004, 47, 2075. doi:10.1021/jm031044w |
| 4. | Cullen, J. J.; Hinkhouse, M. M.; Grady, M.; Gaut, A. W.; Liu, J.; Zhang, Y. P.; Weydert, C. J. D.; Domann, F. E.; Oberley, L. W. Cancer Res. 2003, 63, 5513. |
| 9. | Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi:10.1021/cr068357u |
| 10. | Riduan, S. N.; Zhang, Y. Dalton Trans. 2010, 39, 3347. doi:10.1039/b920163g |
| 11. | Huang, K.; Sun, C.-L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435. doi:10.1039/c0cs00129e |
| 12. | Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W. A.; Kühn, F. E. Angew. Chem., Int. Ed. 2011, 50, 8510. doi:10.1002/anie.201102010 |
| 13. | Zhang, W.; Lü, X. Chin. J. Catal. 2012, 33, 745. doi:10.1016/S1872-2067(11)60390-2 |
| 14. | Tsuji, Y.; Fujihara, T. Chem. Commun. 2012, 48, 9956. doi:10.1039/c2cc33848c |
| 15. | Zhang, L.; Hou, Z. Chem. Sci. 2013, 4, 3395. doi:10.1039/c3sc51070k |
| 16. | Aresta, M.; Dibenedetto, A.; Angelini, A. Chem. Rev. 2014, 114, 1709. doi:10.1021/cr4002758 |
© 2015 Zhang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)