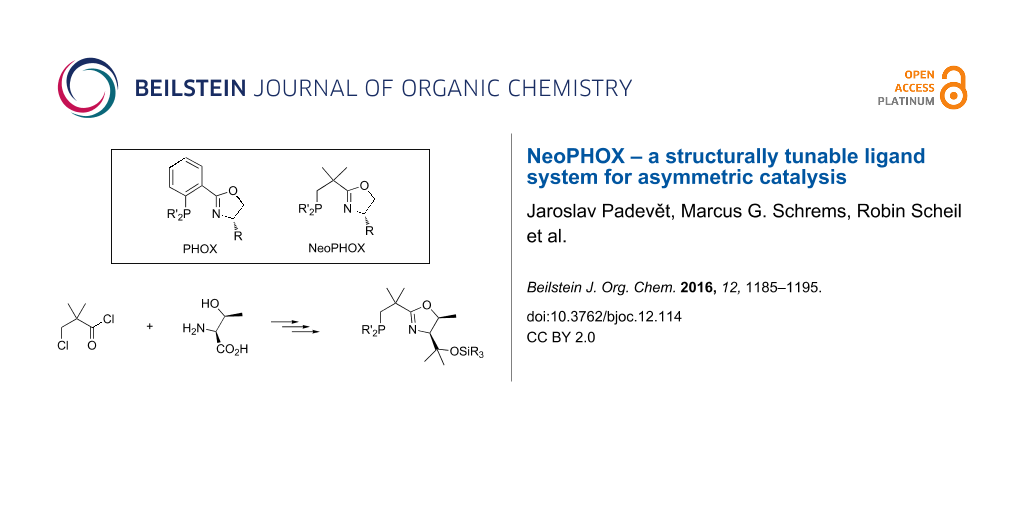
Abstract
A synthesis of new NeoPHOX ligands derived from serine or threonine has been developed. The central intermediate is a NeoPHOX derivative bearing a methoxycarbonyl group at the stereogenic center next to the oxazoline N atom. The addition of methylmagnesium chloride leads to a tertiary alcohol, which can be acylated or silylated to produce NeoPHOX ligands with different sterical demand. The new NeoPHOX ligands were tested in the iridium-catalyzed asymmetric hydrogenation and palladium-catalyzed allylic substitution. In both reactions high enantioselectivities were achieved, that were comparable to the enantioselectivities obtained with the up to now best NeoPHOX ligand derived from expensive tert-leucine.

Graphical Abstract
Introduction
Since their introduction and first successful application in enantioselective palladium-catalyzed allylic substitution in 1993 [1-3], chiral phosphinooxazolines (PHOX ligands) have emerged as a widely used privileged ligand class [4-12]. One of the major areas of application of PHOX and related N,P ligands is the iridium-catalyzed asymmetric hydrogenation [13-16]. Compared to rhodium and ruthenium catalysts, iridium complexes derived from chiral N,P ligands show a much broader substrate scope in the hydrogenation of olefins, as they do not require any coordinating groups in the substrate.
Although high enantioselectivities were achieved with the initially developed Ir-PHOX catalysts, the range of olefins that gave satisfactory results was limited. Consequently, a variety of other ligand classes derived from oxazolines or other nitrogen heterocycles were explored and eventually, several highly effective ligand systems were found that have considerably enhanced the range of functionalized and unfunctionalized olefins that can be hydrogenated with high efficiency and excellent enantioselectivity. Among the many oxazoline-derived ligands that have been reported in the literature, SimplePHOX [17] and ThrePHOX [18] have emerged as some of the most versatile and most easily accessible ligand classes. However, although iridium complexes derived from these ligands are air and moisture-stable, the free ligands are prone to hydrolysis and oxidation. Especially SimplePHOX ligands, although they are readily prepared from amino alcohols in just two steps, suffer from these problems resulting in low yields and difficult purification steps.
We therefore decided to explore structural analogs, in which the P–O bond had been replaced by a P–C bond [19]. We hoped that such ligands, which we named NeoPHOX because of the neopentyl backbone, would be as easily accessible as SimplePHOX but more stable against air and moisture and fulfill the criteria for a scalable high yielding synthesis (Figure 1).
![[1860-5397-12-114-1]](/bjoc/content/figures/1860-5397-12-114-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structural motifs of phospinooxazoline ligands.
Figure 1: Structural motifs of phospinooxazoline ligands.
Results and Discussion
1st Generation NeoPHOX ligands
A potential very short route to NeoPHOX ligands starting from commercially available 3-chloropivaloyl chloride (3) is shown in Scheme 1. Although nucleophilic substitutions at the neopentyl carbon atoms are known to be difficult, literature precedence indicated that the introduction of the phosphine group might be possible by the reaction of a diarylphosphide anion with chloroalkyloxazoline 2.
![[1860-5397-12-114-i1]](/bjoc/content/inline/1860-5397-12-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis for NeoPHOX ligands.
Scheme 1: Retrosynthetic analysis for NeoPHOX ligands.
Ashby et al. [20] investigated the reactivity of neopentyl halide systems with various metal diphenylphosphides and found that the reaction indeed took place. While for iodides evidence for a radical single electron transfer process as the major pathway was found, the corresponding bromides and chlorides seemed to prefer an SN2 mechanism. Rossi et al. [21] showed that substitution products can be obtained in good yields from neopentyl chloride derivatives and sodium diphenylphosphide in liquid ammonia by an SNR2 reaction induced by UV irradiation. Huttner and co-workers [22] on the other hand achieved high yields in nucleophilic substitutions of diarylphosphines with MeC(CH2Cl)3 in DMSO using aqueous KOH as base at elevated temperature but without irradiation.
In initial model studies using neopentyl chloride and a commercially available solution of KPPh2 in THF we obtained high yields of neopentyldiphenylphosphine under reflux conditions [19]. Encouraged by these results, a series of chloroalkyl-substituted oxazolines 2 were prepared from 3-chloropivaloyl chloride (3) by amide formation and subsequent cyclization with the Burgess reagent (methyl N-(triethylammoniumsulfonyl)carbamate) using standard procedures (Scheme 2). Subsequent nucleophilic substitution with KPP2 in refluxing THF proved to be much faster than the analogous reaction with neopentyl chloride. After 6 hours, the KPPh2 was fully converted to the desired phosphinooxazoline, as evidenced by the 31P NMR spectrum, which showed only the product signal. The reaction also proceeded well with other diarylphosphides such as KP(o-Tol)2 or KP(3,5-MePh)2 prepared in situ from the secondary phosphines by deprotonation with KH in refluxing THF. The NeoPHOX ligands, which were obtained by this route in high overall yield, proved to be air and moisture stable and could be obtained in analytically pure form by simple filtration through silica gel [19]. Complexation with [Ir(COD)Cl]2 and subsequent anion exchange with NaBArF (sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate) led to the corresponding iridium precatalysts in high yields. These complexes as well showed high stability against oxygen and moisture and could be stored in air for several months without notable decomposition.
![[1860-5397-12-114-i2]](/bjoc/content/inline/1860-5397-12-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1st generation NeoPHOX Ir-complexes [19].
Scheme 2: Synthesis of 1st generation NeoPHOX Ir-complexes [19].
As originally assumed, the NeoPHOX complexes Ir-1a–e turned out to be highly efficient catalysts. They induced similar and in some cases even higher enantioselectivities compared to the analogous SimplePHOX complexes, as shown by the results obtained in the hydrogenation of a range of test substrates (Figure 2) [19]. Considering the more practicable synthesis, which is well suited for large-scale preparation, and the higher stability of the free ligands, NeoPHOX ligands provide a superior alternative to the SimplePHOX ligands. The only drawback these compounds share with many state-of-the-art oxazoline-based ligands is the high cost of tert-leucinol as starting material, which reduces their potential for industrial-scale application. We therefore thought of ways to replace the tert-butyloxazoline ring by an equally effective oxazoline unit derived from a less expensive precursor.
![[1860-5397-12-114-2]](/bjoc/content/figures/1860-5397-12-114-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Asymmetric hydrogenation with iridium-NeoPHOX catalysts [19].
Figure 2: Asymmetric hydrogenation with iridium-NeoPHOX catalysts [19].
An option, which had been previously used in the development of PHOX ligands [23-27], is to replace the tert-butyl group by an isopropyl group and at the same time introducing two bulky substituents at C(5) (see Figure 3). Ligands of this type are accessible from valine, which is much less expensive than tert-leucine. The steric hindrance exerted by the geminal substituents at C(5) is expected to direct the isopropyl methyl groups toward the coordination sphere, creating a steric environment which resembles that of a tert-butyloxazoline ligand.
![[1860-5397-12-114-3]](/bjoc/content/figures/1860-5397-12-114-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Employing L-valine as a starting material for C5 substituted oxazoline.
Figure 3: Employing L-valine as a starting material for C5 substituted oxazoline.
To test the viability of this approach, we synthesized the valine-derived NeoPHOX ligand 10 with two geminal phenyl substituents at C(5) according to the route shown in Scheme 3.
![[1860-5397-12-114-i3]](/bjoc/content/inline/1860-5397-12-114-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of a C(5)-disubstituted NeoPHOX-Ir complex.
Scheme 3: Synthesis of a C(5)-disubstituted NeoPHOX-Ir complex.
Unfortunately, the iridium complex Ir-10 prepared from this ligand proved to be an inefficient catalyst. The enantioselectivities in the hydrogenation of various test substrates were much lower than those induced by the tert-butyloxazoline analog with the exception of the result obtained with the allylic alcohol S2 (Table 1). Surprisingly, the presence of two geminal phenyl substituents at C(5) had a negative impact on the enantioselectivity, as shown by the substantially higher ee values induced by the analogous catalyst Ir-1a [28] lacking substituents at C(5).
Table 1: Hydrogenation results employing 1st generation NeoPHOX catalysts.a
|
S1
ee [%] yield [%] |
S5
ee [%] yield [%] |
S2
ee [%] yield [%] |
S3
ee [%] yield [%] |
|
|---|---|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-114-i11.svg?max-width=637&scale=1.0)
Ir-10 |
19 (R)
93 |
35 (S)
>99 |
84 (–)
>99 |
63 (R)
41 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-114-i12.svg?max-width=637&scale=1.0)
Ir-1a |
74 (R)
>99 |
53 (S)
89 |
88 (–)
>99 |
85 (R)
>99 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-114-i13.svg?max-width=637&scale=1.0)
Ir-1b |
97 (R)
>99 |
92 (S)
>99 |
83 (–)
>99 |
95 (R)
>99 |
aReaction conditions: 50 bar, 2 h, 1 mol % catalyst, 0.1 mmol substrate, 0.5 mL CH2Cl2.
Based on these negative results we chose a new approach based on serine or threonine as starting materials (Figure 4). The carboxyl group of these amino acids serves as surrogate for the tert-butyl group of tert-leucine, which can be converted to a sterically demanding substituent by double addition of Grignard or alkyllithium reagents and subsequent protection of the resulting tertiary alcohol. An attractive feature of this approach is that by proper choice of the alkylmetal reagent and the protecting group, the steric properties of the ligand can be optimized for a specific application. PHOX ligands of this type have been previously prepared from serine and successfully used in iridium-catalyzed hydrogenation [29,30].
![[1860-5397-12-114-4]](/bjoc/content/figures/1860-5397-12-114-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Retrosynthetic analysis for NeoPHOX ligands derived from serine and threonine.
Figure 4: Retrosynthetic analysis for NeoPHOX ligands derived from serine and threonine.
Synthesis of serine- and threonine-derived NeoPHOX ligands
The synthesis that led to the NeoPHOX ligand 14 in 46% overall yield is shown in Scheme 4. In the first step threonine methyl ester hydrochloride was converted to the desired amide 11 in 96% yield. Subsequent cyclization with the Burgess reagent afforded the oxazoline 12 in 89% yield with clean inversion of configuration at the hydroxy-substituted carbon atom. Double addition of methylmagnesium chloride and subsequent introduction of the diphenylphosphine unit proceeded well in 68% yield following the procedures worked out for the 1st generation NeoPHOX ligands.
![[1860-5397-12-114-i4]](/bjoc/content/inline/1860-5397-12-114-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Revisited synthetic strategy for the preparation of a threonine-based NeoPHOX ligand.
Scheme 4: Revisited synthetic strategy for the preparation of a threonine-based NeoPHOX ligand.
Alternative strategies involving Grignard addition to the threonine methyl ester or N-protected derivatives were also briefly investigated, but failed. Grignard addition to the chloro amide 11 was not attempted because earlier studies with chloro amide 15 had shown that an undesired β-lactam 16 was formed upon treatment with Grignard reagents (Scheme 5) [28].
![[1860-5397-12-114-i5]](/bjoc/content/inline/1860-5397-12-114-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
All attempts to prepare analogous serine-derived NeoPHOX ligands by the established route for the synthesis of the threonine-derived NeoPHOX ligand 14 led to unexpected problems (Scheme 6). Oxazoline formation with the Burgess reagent did not produce the desired oxazoline (mixture of unidentified products). However, eventually an alternative method using diethylaminosulfurtrifluoride (DAST) [31] was found that afforded the chloroalkyloxazoline 18 in 95% yield after short reaction times at −78 °C.
![[1860-5397-12-114-i6]](/bjoc/content/inline/1860-5397-12-114-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthetic strategy for the synthesis of the serine-derived NeoPHOX ligand.
Scheme 6: Synthetic strategy for the synthesis of the serine-derived NeoPHOX ligand.
Treatment of this product with methylmagnesium chloride under the same conditions used for the threonine derivative 12 (Scheme 4), led to an unidentified mixture of products. Surprisingly, when the Grignard reagent was added at 0 °C instead of −78 °C and reaction mixture subsequently warmed up to room temperature, the desired product 19 was formed in 78% yield. After having resolved these issues, the desired serine-based NeoPHOX ligand 20 was obtained in 23% overall yield from serine methylester hydrochloride.
Next we converted the tertiary alcohols 14 and 20 to a range of silylated and acetylated derivatives in order to evaluate the influence of sterically and electronically different substituents on the enantioselectivity and catalytic activity of the corresponding iridium complexes. Initial attempts to introduce a silyl or acetyl group by deprotonation with potassium hydride and subsequent reaction with silyl triflates or acetyl chloride failed. The NeoPHOX ligand 14 showed no reaction under these conditions, although this method had been successfully used for the alkylation and acylation of the tertiary alcohol function of analogous serine derived PHOX ligands [30]. Among various amines, 2,6-lutidine was finally identified as a suitable base for conversion to the desired derivatives in 67–76% yields (Scheme 7). From these ligands a library of iridium catalysts was prepared by complexation with bis(1,5-cyclooctadiene)diiridium(I) dichloride, followed by anion exchange with NaBArF.
![[1860-5397-12-114-i7]](/bjoc/content/inline/1860-5397-12-114-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Derivatization of the 2nd generation NeoPHOX ligands and formation of their iridium complexes.
Scheme 7: Derivatization of the 2nd generation NeoPHOX ligands and formation of their iridium complexes.
Comparative hydrogenation studies with iridium catalysts derived from 1st and 2nd generation NeoPHOX ligands and serine-based PHOX ligands
A set of seven Ir-NeoPHOX catalysts was evaluated in the asymmetric hydrogenation of three alkenes and acetophenone phenylimine (Table 2). For comparison, the results previously reported for four related serine-based Ir-PHOX complexes are included in the table. Overall, the tert-leucine-derived complex Ir-1b and complexes Ir-14-TBDMS and Ir-20-TBDMS with a bulky tert-butyldimethylsilyloxy group showed the highest reactivity, resulting in full conversion of all three olefins. On the other hand catalysts Ir-14-OH and ser-PHOX-OMe bearing a free hydroxy and a methoxy group, respectively, gave very low or no conversion. In the case of complex ser-PHOX-OMe [30] it was found that the methoxy function in the oxazoline ring induced deactivation of the catalyst due to coordination to the metal center resulting in a stable hydride-bridged dinuclear complex under hydrogenation conditions. The analogous catalysts Ir-14-OAc and ser-PHOX-OAc with an acyloxy substituent proved to be more reactive, however, conversion still remained incomplete with olefins S1 and S3. Similar results were obtained with the analogous NeoPHOX complex Ir-14-OAc bearing an acetoxy group at the oxazoline ring. Apparently, a polar potentially coordinating substituent near the metal center reduces or can even completely inhibit catalytic activity. The deactivating effect of a coordinating oxygen function could also explain the different conversions observed with the silyl ether-bearing catalysts, decreasing in the series t-BuMe2Si > Et3Si > Me3Si. It has been shown that Ir-hydride complexes that are formed as intermediates during hydrogenation are strong Brønsted acids [32], which can cause cleavage of trimethylsilyl ethers under hydrogenation conditions [33]. So partial desilylation liberating a free hydroxy group during hydrogenation may well be the reason for the low conversion obtained with catalyst Ir-14-TMS bearing an acid-labile trimethylsilyl ether group.
Table 2: Hydrogenation of standard substrates using serine- and threonine-derived NeoPHOX catalysts.
|
S1
ee [%] yield [%] |
S2
ee [%] yield [%] |
S3
ee [%] yield [%] |
S4
ee [%] yield [%] |
|
|---|---|---|---|---|
| Ir-14-OH | n.d. |
41 (+)
10 |
n.d. |
7 (S)
4 |
| Ir-14-OAc |
87 (S)
59 |
90 (+)
92 |
90 (S)
>44 |
16 (S)
73 |
| Ir-14-TMS |
92 (S)
38 |
88 (+)
82 |
82 (S)
>40 |
67 (S)
5 |
| Ir-14-TES |
97/b96 (S)
59/b87 |
89/b88 (+)
60/b>99 |
90/b90 (S)
87/b98 |
68/b62 (S)
8/b32 |
| Ir-14-TBDMS |
96 (S)
>99 |
91 (+)
>99 |
94 (S)
>99 |
49 (S)
79 |
| Ir-20-TBDMS |
90 (S)
>99 |
84 (+)
>99 |
92 (S)
>99 |
77 (S)
79 |
| Ir-1b |
97 (R)
>99 |
97% (-)
>99 |
97% (R)
>99 |
97 (R)
>99 |
| ser-PHOX-OMe [30] |
n.d.
1 |
57 (+)
9 |
32 (S)
3 |
5 (R)
2 |
| ser-PHOX-Bn [30] |
92 (S)
27 |
79 (+)
43 |
46 (S)
34 |
21 (S)
1 |
| ser-PHOX-OAc [30] |
92 (S)
90 |
91 (+)
>99 |
56 (S)
70 |
56 (R)
99 |
| ser-PHOX-Bz [30] |
92(S)
88 |
92 (+)
>99 |
69 (S)
91 |
53 (R)
>99 |
aReaction conditions: 50 bar, 2h, 1 mol % catalyst, 0.1 mmol substrate, 0.5 mL CH2Cl2; breaction time 16 h; ![[Graphic 4]](/bjoc/content/inline/1860-5397-12-114-i14.svg?max-width=637&scale=1.18182)
Among the NeoPHOX catalysts, the most reactive complex with a bulky TBDMS group showed also the highest enantioselectivities in the hydrogenation of alkenes S1–3. It also outperformed the most efficient serine-derived PHOX catalysts ser-PHOX-OAc and ser-PHOX-OBz. In terms of activity and enantiomeric excess the performance of this catalyst was comparable to the tert-leucine-derived complex Ir-1b. The analogous serine-based complex Ir-20-TBDMS gave somewhat lower enantioselectivities. In the hydrogenation of imine S4 none of the catalysts shown in Table 2 induced high enantioselectivity.
Crystal structures of iridium NeoPHOX complexes
The three dimensional structures of several threonine and serine-derived NeoPHOX iridium complexes were determined by X-ray diffraction and compared with known phosphinooxazoline complexes. We were interested to see whether the differences in steric shielding of the coordination sphere by the protecting groups on the tertiary alcohol function correlated with the hydrogenation results. From the obtained crystal structures, it can be seen that the conformations of the threonine and serine-derived NeoPHOX iridium complexes are essentially the same as those of the tert-leucine-derived NeoPHOX complex with respect to the geometry of the 6-membered iridacycle, which retains a boat-shaped conformation (Figure 5). No notable divergence in the steric environment of the iridium center in the four silyloxy-bearing NeoPHOX complexes could be observed that would explain the difference in enantioselectivity induced by these catalysts.
![[1860-5397-12-114-5]](/bjoc/content/figures/1860-5397-12-114-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Crystal structures of selected Ir-complexes. Hydrogen atoms, COD and BArF anions were omitted for clarity.
Figure 5: Crystal structures of selected Ir-complexes. Hydrogen atoms, COD and BArF anions were omitted for c...
In contrast to the methoxy-bearing serine-derived PHOX complex ser-PHOX-OMe, the protected tertiary alcohol group in these complexes does not point towards the iridium center. The interaction of the ether oxygen atom with the iridium center was the reason for the lack of reactivity of this serine-based PHOX complex. In case of the silyl ethers the oxygen atom is strongly shielded preventing coordination to the iridium center.
Palladium-catalyzed allylic substitution
As phosphinooxazoline ligands were originally designed for asymmetric palladium-catalyzed allylic substitutions, we tested the new NeoPHOX ligands in this reaction as well. For comparison with state-of-the-art ligands, we chose the standard test reaction of (E)-1,3-diphenylallylacetate with dimethyl malonate as nucleophile (Scheme 8).
![[1860-5397-12-114-i8]](/bjoc/content/inline/1860-5397-12-114-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Asymmetric palladium-catalyzed allylic substitution with rac-(E)-1,3-diphenylallyl acetate.
Scheme 8: Asymmetric palladium-catalyzed allylic substitution with rac-(E)-1,3-diphenylallyl acetate.
The two catalysts derived from ligand 14 with a free hydroxy group and the corresponding triethylsilyl-protected derivative both performed well affording enantioselectivities of 90% and 98% ee, respectively. Ligand 14-TES was even more effective than the corresponding tert-leucine-derived NeoPHOX ligand 1b (97% ee).
Next, we also tested (E)-1,3-dimethylallyl acetate as substrate (Scheme 9). All three tested catalysts gave disappointing results. The yield was low in all cases and the enantiomeric excess reached only 41% for the best ligand 14. Interestingly, the sterically more demanding ligand 14-TES did not lead to higher enantioselectivity as for 1,3-diphenylallyl acetate.
![[1860-5397-12-114-i9]](/bjoc/content/inline/1860-5397-12-114-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Asymmetric palladium-catalyzed allylic substitution with rac-(E)-1,3-dimethylallyl acetate.
Scheme 9: Asymmetric palladium-catalyzed allylic substitution with rac-(E)-1,3-dimethylallyl acetate.
Finally, the ligands were also tested on a more demanding cyclic substrate (Scheme 10). In the allylic substitution with cyclohex-2-en-1-yl benzoate a notable enantiomeric excess of 70% was achieved using ligand 14-TES, in striking contrast to the extremely low enantioselectivities reported for analogous PHOX ligands [4]. Using ligand 14 with a free hydroxy group the result was less satisfying. Not only the yield dropped to 53% but also the ee was substantially lower than with the triethylsilyl analog 14-TES. For NeoPHOX ligand 1b the enantiomeric excess was even lower (5%). These results demonstrate that the 2nd generation NeoPHOX ligands possess potential for palladium-catalyzed allylic substitutions.
![[1860-5397-12-114-i10]](/bjoc/content/inline/1860-5397-12-114-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Asymmetric palladium-catalyzed allylic substitution with a cyclic substrate.
Scheme 10: Asymmetric palladium-catalyzed allylic substitution with a cyclic substrate.
Conclusion
The most successful, most widely applied oxazoline-based N,P ligands are derived from the unnatural amino acid tert-leucine. The high price of this starting material is an impeding factor for large-scale applications. The NeoPHOX ligands derived from threonine provide an attractive alternative in this respect. Threonine as a chiral building block is available in both enantiomeric forms at a moderate price. The most effective ligand in this series, bearing a bulky CMe2OSiMe2t-Bu group at the stereogenic center, induced excellent enantioselectivities in the Ir-catalyzed asymmetric hydrogenation of olefins, with ee values in the same range as those reported for the best N,P ligands including the tert-leucine-derived NeoPHOX analog. In the asymmetric Pd-catalyzed allylic substitution as well, promising enantioselectivities were obtained, indicating a considerable potential for this ligand class in asymmetric catalysis.
A further notable feature of these ligands is their flexible synthesis, which allows easy variation of the substituent at the stereogenic center. Using different Grignard reagents in the addition to the ester group of a late stage intermediate, an array of tertiary alcohol derivatives is available. The subsequent silylation or acylation of the hydroxy group gives access to a library of diverse NeoPHOX ligands. In this way the steric size and the coordination ability of the substituent at the stereogenic center can be tuned for a specific application.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of all compounds and copies of 1H and 13C NMR spectra of selected molecules. | ||
| Format: PDF | Size: 1.9 MB | Download |
References
-
von Matt, P.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 566–568. doi:10.1002/anie.199305661
Return to citation in text: [1] -
Sprinz, J.; Helmchen, G. Tetrahedron Lett. 1993, 34, 1769–1772. doi:10.1016/S0040-4039(00)60774-8
Return to citation in text: [1] -
Dawson, G. J.; Frost, C. G.; Williams, J. M. J.; Coote, S. J. Tetrahedron Lett. 1993, 34, 3149–3150. doi:10.1016/S0040-4039(00)93403-8
Return to citation in text: [1] -
Helmchen, G.; Pfaltz, A. Acc. Chem. Res. 2000, 33, 336–345. doi:10.1021/ar9900865
Return to citation in text: [1] [2] -
Linton, E. C.; Kozlowski, M. C. J. Am. Chem. Soc. 2008, 130, 16162–16163. doi:10.1021/ja807026z
Return to citation in text: [1] -
Wang, Y.; Liu, D.; Meng, Q.; Zhang, W. Tetrahedron: Asymmetry 2009, 20, 2510–2512. doi:10.1016/j.tetasy.2009.10.015
Return to citation in text: [1] -
Petersen, K. S.; Stoltz, B. M. Tetrahedron 2011, 67, 4352–4357. doi:10.1016/j.tet.2011.04.046
Return to citation in text: [1] -
Seto, M.; Roisen, J. L.; Stoltz, B. M. Angew. Chem., Int. Ed. 2008, 47, 6873–6876. doi:10.1002/anie.200801424
Return to citation in text: [1] -
Marinescu, S. C.; Nishimata, T.; Mohr, J. T.; Stoltz, B. M. Org. Lett. 2008, 10, 1039–1042. doi:10.1021/ol702821j
Return to citation in text: [1] -
Carroll, M. P.; Guiry, P. J. Chem. Soc. Rev. 2014, 43, 819–833. doi:10.1039/C3CS60302D
Return to citation in text: [1] -
Marziale, A. N.; Duquette, D. C.; Craig, R. A., II; Kim, K. E.; Liniger, M.; Numajiri, Y.; Stoltz, B. M. Adv. Synth. Catal. 2015, 357, 2238–2245. doi:10.1002/adsc.201500253
Return to citation in text: [1] -
Zirimwabagabo, J.-O.; Harrity, J. P. A. Chem. Commun. 2014, 50, 2735–2737. doi:10.1039/c4cc00109e
Return to citation in text: [1] -
Verendel, J. J.; Pàmies, O.; Diéguez, M.; Andersson, P. G. Chem. Rev. 2014, 114, 2130–2169. doi:10.1021/cr400037u
Return to citation in text: [1] -
Woodmansee, D. H.; Pfaltz, A. Top. Organomet. Chem. 2011, 34, 31–76. doi:10.1007/978-3-642-15334-1_3
Return to citation in text: [1] -
Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402–1411. doi:10.1021/ar700113g
Return to citation in text: [1] -
Cui, X.; Burgess, K. Chem. Rev. 2005, 105, 3272–3296. doi:10.1021/cr0500131
Return to citation in text: [1] -
Smidt, S. P.; Menges, F.; Pfaltz, A. Org. Lett. 2004, 6, 2023–2026. doi:10.1021/ol049235w
Return to citation in text: [1] -
Menges, F.; Pfaltz, A. Adv. Synth. Catal. 2002, 344, 40–44. doi:10.1002/1615-4169(200201)344:1<40::AID-ADSC40>3.0.CO;2-7
Return to citation in text: [1] -
Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Ashby, E. C.; Gurumurthy, R.; Ridlehuber, R. W. J. Org. Chem. 1993, 58, 5832–5837. doi:10.1021/jo00073a051
Return to citation in text: [1] -
Duca, J. S., Jr.; Gallego, M. H.; Pierini, A. B.; Rossi, R. A. J. Org. Chem. 1999, 64, 2626–2629. doi:10.1021/jo980657r
Return to citation in text: [1] -
Muth, A.; Walter, O.; Huttner, G.; Asam, A.; Zsolnai, L.; Emmerich, C. J. Organomet. Chem. 1994, 468, 149–163. doi:10.1016/0022-328X(94)80044-8
Return to citation in text: [1] -
Stohler, R.; Wahl, F.; Pfaltz, A. Synthesis 2005, 1431–1436. doi:10.1055/s-2005-865313
Return to citation in text: [1] -
Stohler, R. Asymmetric Metal-Catalyzed [3 + 2] Cycloadditions of Azomethine Ylides. Ph.D. Thesis, University of Basel, Switzerland, 2006.
http://edoc.unibas.ch/605/1/DissB_7907.pdf
Return to citation in text: [1] -
Bélanger, E.; Pouliot, M.-F.; Paquin, J.-F. Org. Lett. 2009, 11, 2201–2204. doi:10.1021/ol9005618
Return to citation in text: [1] -
Bélanger, E.; Pouliot, M.-F.; Courtemanche, M.-A.; Paquin, J.-F. J. Org. Chem. 2012, 77, 317–331. doi:10.1021/jo2019653
Return to citation in text: [1] -
Liu, W.-B.; Reeves, C. M.; Virgil, S. C.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 10626–10629. doi:10.1021/ja4052075
Return to citation in text: [1] -
Schrems, M. G. Iridium-katalysierte Hydrierung unfunktionalisierter Olefine. Ph.D. Thesis, University of Basel, Switzerland, 2009.
http://edoc.unibas.ch/1007/1/Dissertation_Marcus_Schrems.pdf.
Return to citation in text: [1] [2] -
Franzke, A.; Pfaltz, A. Chem. – Eur. J. 2011, 17, 4131–4144. doi:10.1002/chem.201003314
Return to citation in text: [1] -
Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Phillips, A. J.; Uto, Y.; Wipf, P.; Reno, M. J.; Williams, D. R. Org. Lett. 2000, 2, 1165–1168. doi:10.1021/ol005777b
Return to citation in text: [1] -
Zhu, Y.; Fan, Y.; Burgess, K. J. Am. Chem. Soc. 2010, 132, 6249–6253. doi:10.1021/ja101233g
Return to citation in text: [1] -
Schumacher, A.; Bernasconi, M.; Pfaltz, A. Angew. Chem., Int. Ed. 2013, 52, 7422–7425. doi:10.1002/anie.201301251
Return to citation in text: [1]
| 4. | Helmchen, G.; Pfaltz, A. Acc. Chem. Res. 2000, 33, 336–345. doi:10.1021/ar9900865 |
| 1. | von Matt, P.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 566–568. doi:10.1002/anie.199305661 |
| 2. | Sprinz, J.; Helmchen, G. Tetrahedron Lett. 1993, 34, 1769–1772. doi:10.1016/S0040-4039(00)60774-8 |
| 3. | Dawson, G. J.; Frost, C. G.; Williams, J. M. J.; Coote, S. J. Tetrahedron Lett. 1993, 34, 3149–3150. doi:10.1016/S0040-4039(00)93403-8 |
| 18. | Menges, F.; Pfaltz, A. Adv. Synth. Catal. 2002, 344, 40–44. doi:10.1002/1615-4169(200201)344:1<40::AID-ADSC40>3.0.CO;2-7 |
| 23. | Stohler, R.; Wahl, F.; Pfaltz, A. Synthesis 2005, 1431–1436. doi:10.1055/s-2005-865313 |
| 24. |
Stohler, R. Asymmetric Metal-Catalyzed [3 + 2] Cycloadditions of Azomethine Ylides. Ph.D. Thesis, University of Basel, Switzerland, 2006.
http://edoc.unibas.ch/605/1/DissB_7907.pdf |
| 25. | Bélanger, E.; Pouliot, M.-F.; Paquin, J.-F. Org. Lett. 2009, 11, 2201–2204. doi:10.1021/ol9005618 |
| 26. | Bélanger, E.; Pouliot, M.-F.; Courtemanche, M.-A.; Paquin, J.-F. J. Org. Chem. 2012, 77, 317–331. doi:10.1021/jo2019653 |
| 27. | Liu, W.-B.; Reeves, C. M.; Virgil, S. C.; Stoltz, B. M. J. Am. Chem. Soc. 2013, 135, 10626–10629. doi:10.1021/ja4052075 |
| 17. | Smidt, S. P.; Menges, F.; Pfaltz, A. Org. Lett. 2004, 6, 2023–2026. doi:10.1021/ol049235w |
| 28. |
Schrems, M. G. Iridium-katalysierte Hydrierung unfunktionalisierter Olefine. Ph.D. Thesis, University of Basel, Switzerland, 2009.
http://edoc.unibas.ch/1007/1/Dissertation_Marcus_Schrems.pdf. |
| 13. | Verendel, J. J.; Pàmies, O.; Diéguez, M.; Andersson, P. G. Chem. Rev. 2014, 114, 2130–2169. doi:10.1021/cr400037u |
| 14. | Woodmansee, D. H.; Pfaltz, A. Top. Organomet. Chem. 2011, 34, 31–76. doi:10.1007/978-3-642-15334-1_3 |
| 15. | Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402–1411. doi:10.1021/ar700113g |
| 16. | Cui, X.; Burgess, K. Chem. Rev. 2005, 105, 3272–3296. doi:10.1021/cr0500131 |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 4. | Helmchen, G.; Pfaltz, A. Acc. Chem. Res. 2000, 33, 336–345. doi:10.1021/ar9900865 |
| 5. | Linton, E. C.; Kozlowski, M. C. J. Am. Chem. Soc. 2008, 130, 16162–16163. doi:10.1021/ja807026z |
| 6. | Wang, Y.; Liu, D.; Meng, Q.; Zhang, W. Tetrahedron: Asymmetry 2009, 20, 2510–2512. doi:10.1016/j.tetasy.2009.10.015 |
| 7. | Petersen, K. S.; Stoltz, B. M. Tetrahedron 2011, 67, 4352–4357. doi:10.1016/j.tet.2011.04.046 |
| 8. | Seto, M.; Roisen, J. L.; Stoltz, B. M. Angew. Chem., Int. Ed. 2008, 47, 6873–6876. doi:10.1002/anie.200801424 |
| 9. | Marinescu, S. C.; Nishimata, T.; Mohr, J. T.; Stoltz, B. M. Org. Lett. 2008, 10, 1039–1042. doi:10.1021/ol702821j |
| 10. | Carroll, M. P.; Guiry, P. J. Chem. Soc. Rev. 2014, 43, 819–833. doi:10.1039/C3CS60302D |
| 11. | Marziale, A. N.; Duquette, D. C.; Craig, R. A., II; Kim, K. E.; Liniger, M.; Numajiri, Y.; Stoltz, B. M. Adv. Synth. Catal. 2015, 357, 2238–2245. doi:10.1002/adsc.201500253 |
| 12. | Zirimwabagabo, J.-O.; Harrity, J. P. A. Chem. Commun. 2014, 50, 2735–2737. doi:10.1039/c4cc00109e |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 22. | Muth, A.; Walter, O.; Huttner, G.; Asam, A.; Zsolnai, L.; Emmerich, C. J. Organomet. Chem. 1994, 468, 149–163. doi:10.1016/0022-328X(94)80044-8 |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 21. | Duca, J. S., Jr.; Gallego, M. H.; Pierini, A. B.; Rossi, R. A. J. Org. Chem. 1999, 64, 2626–2629. doi:10.1021/jo980657r |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 20. | Ashby, E. C.; Gurumurthy, R.; Ridlehuber, R. W. J. Org. Chem. 1993, 58, 5832–5837. doi:10.1021/jo00073a051 |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 19. | Schrems, M. G.; Pfaltz, A. Chem. Commun. 2009, 6210–6212. doi:10.1039/b912680e |
| 31. | Phillips, A. J.; Uto, Y.; Wipf, P.; Reno, M. J.; Williams, D. R. Org. Lett. 2000, 2, 1165–1168. doi:10.1021/ol005777b |
| 29. | Franzke, A.; Pfaltz, A. Chem. – Eur. J. 2011, 17, 4131–4144. doi:10.1002/chem.201003314 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 28. |
Schrems, M. G. Iridium-katalysierte Hydrierung unfunktionalisierter Olefine. Ph.D. Thesis, University of Basel, Switzerland, 2009.
http://edoc.unibas.ch/1007/1/Dissertation_Marcus_Schrems.pdf. |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 32. | Zhu, Y.; Fan, Y.; Burgess, K. J. Am. Chem. Soc. 2010, 132, 6249–6253. doi:10.1021/ja101233g |
| 33. | Schumacher, A.; Bernasconi, M.; Pfaltz, A. Angew. Chem., Int. Ed. 2013, 52, 7422–7425. doi:10.1002/anie.201301251 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
| 30. | Franzke, A.; Voss, F.; Pfaltz, A. Tetrahedron 2011, 67, 4358–4363. doi:10.1016/j.tet.2011.02.021 |
© 2016 Padevět et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)