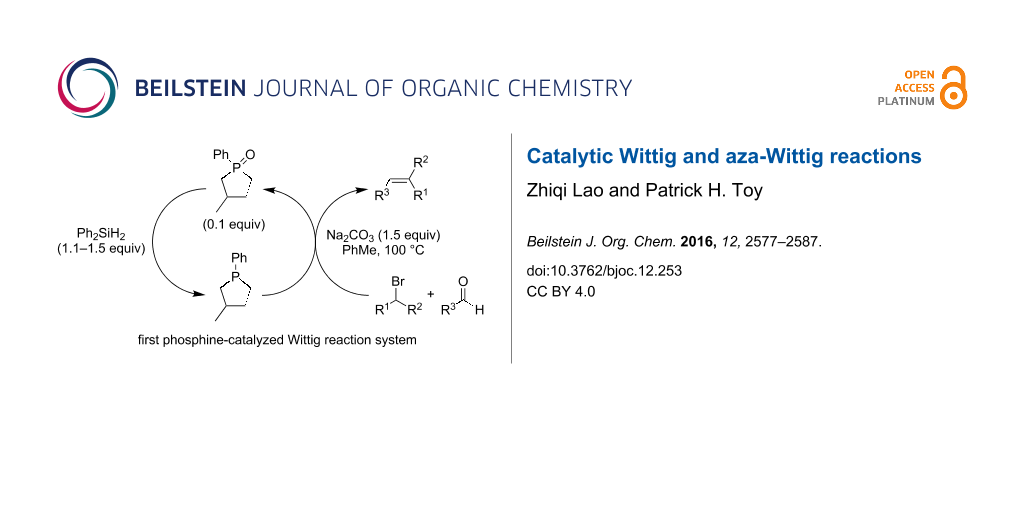
Abstract
This review surveys the literature regarding the development of catalytic versions of the Wittig and aza-Wittig reactions. The first section summarizes how arsenic and tellurium-based catalytic Wittig-type reaction systems were developed first due to the relatively easy reduction of the oxides involved. This is followed by a presentation of the current state of the art regarding phosphine-catalyzed Wittig reactions. The second section covers the field of related catalytic aza-Wittig reactions that are catalyzed by both phosphine oxides and phosphines.

Graphical Abstract
Introduction
The Wittig reaction is a venerable transformation for converting the carbon–oxygen double bond of an aldehyde or a ketone into a carbon–carbon double bond of an alkene group (Scheme 1). Since its introduction over half a century ago [1,2], it has been widely employed in organic synthesis due to its versatility and reliability. The requirement of simple and inexpensive reagents to generate the necessary phosphonium ylide (phosphorane) reactant (a phosphine, typically Ph3P (1), an alkyl halide and a base), also adds to its appeal [3,4]. However, despite its proven utility, the Wittig reaction suffers from limitations that may deter from its use, especially on a large scale, in the context of green sustainable chemistry. For example, it has low atom economy due to its requirement of one molar equivalent of a phosphine reagent, and the formation of a corresponding amount of a phosphine oxide, usually Ph3P=O (2). There is also the associated problem of separating a by-product from the desired product when they are formed in equal molar amounts. These major deficiencies of the Wittig reaction have led to numerous efforts towards developing variations of it which are catalytic in the required phosphine, or a surrogate for it, and this research is the major focus of this review [5-8]. Additionally, analogous catalytic aza-Wittig reactions, in which carbon–nitrogen double bonds of imine groups are formed, will also be discussed in the second section of this review.
![[1860-5397-12-253-i1]](/bjoc/content/inline/1860-5397-12-253-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Prototypical Wittig reaction involving in situ phosphonium salt and phosphonium ylide formation.
Scheme 1: Prototypical Wittig reaction involving in situ phosphonium salt and phosphonium ylide formation.
Review
Catalytic Wittig reactions
A key requirement for versions of the Wittig reaction that are catalytic in phosphine is the selective in situ reduction of the P(V) phosphine oxide byproduct back to the P(III) phosphine in the presence of a reducible aldehyde or ketone substrate, an alkyl halide and a base. Thus, it seems that the challenge in developing catalytic versions of the Wittig reaction distils down to identifying and implementing selective reducing conditions that enables the necessary catalyst redox cycling, yet does not reduce either the starting materials or the desired alkene-containing product.
Arsine and telluride-catalyzed reactions
As phosphine oxides are generally very stable and relatively difficult to reduce, the group of Yao-Zeng Huang used their prior findings that arsonium ylides can participate in Wittig-type reactions. Further they found that arsine oxides can be reduced using much milder reaction conditions compared to phosphine oxides. They developed the first reported catalytic Wittig-type reactions in which Bu3As (3, 0.2 equivalents) was used as the catalyst (Scheme 2) [9,10]. The reaction of 3 with an alkyl halide 4 followed by deprotonation using potassium carbonate generated the corresponding arsonium ylide (5) which, in turn, reacted with an aldehyde substrate 6 to produce the alkene-containing product 7 together with Bu3As=O (8). The byproduct 8 was then reduced in situ using triphenylphosphite to regenerate catalyst 3 for participation in another reaction cycle. Overall, the reaction conditions were quite mild, with the reactions being performed at room temperature with only slight excesses of base and reducing reagent being required. It should be noted that the use of only electron-withdrawing groups activated alkyl halides 4, and that aromatic and aliphatic aldehydes 6 worked well in these reactions to produce products 7 in high yields with predominantly E-configuration.
![[1860-5397-12-253-i2]](/bjoc/content/inline/1860-5397-12-253-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Bu3As-catalyzed Wittig-type reactions.
Scheme 2: Bu3As-catalyzed Wittig-type reactions.
Quite a few years later Yong Tang and co-workers, also of the Shanghai Institute of Organic Chemistry, carried on with this research and extended it by using a combination of Ph3As (9, 0.2 equivalents), Fe(TCP)Cl (10, TCP = tetra(p-chlorophenyl)porphyrinate), and ethyl diazoacetate (11) to generate arsonium ylide 12 for use in biphasic catalytic Wittig-type reactions (Scheme 3) [11]. In these reactions sodium hydrosulfite replaced triphenylphosphite as the reducing reagent to convert the byproduct Ph3As=O (13) back into 9 in the aqueous phase of the reaction mixture in order to make the reactions more environmentally friendly. As was the case in the previous work described above, both aromatic and aliphatic aldehydes 6 were suitable substrates in this reaction system to form products 7.
![[1860-5397-12-253-i3]](/bjoc/content/inline/1860-5397-12-253-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Ph3As-catalyzed Wittig-type reactions using Fe(TCP)Cl and ethyl diazoacetate for arsonium ylide generation.
Scheme 3: Ph3As-catalyzed Wittig-type reactions using Fe(TCP)Cl and ethyl diazoacetate for arsonium ylide gen...
Most recently the Tang research group has reported the use of polymer-supported arsine 14 (0.008–0.04 equivalents) as the catalyst in related reactions (Figure 1) [12]. In this work, 14 was found to be the only arsine examined that was able to effectively catalyze Wittig-type reactions of ketone substrates to produce tri- and tetrasubstituted alkene products in very high yields. For these reactions, which required a higher operating temperature than before (110 °C compared to 80 °C), polymethylhydrosiloxane was used as the reductant, and 14 could be recovered and reused efficiently in numerous reaction cycles without loss of catalytic activity.
![[1860-5397-12-253-1]](/bjoc/content/figures/1860-5397-12-253-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Recyclable polymer-supported arsine for catalytic Wittig-type reactions.
Figure 1: Recyclable polymer-supported arsine for catalytic Wittig-type reactions.
At about the time that Huang and co-workers reported their catalytic reactions using 3 (Scheme 2), they also disclosed that Bu2Te (15) could function similarly as a catalyst in such Wittig-type reactions due to the relatively weak tellurium–oxygen bond of dialkyl telluroxides, such as Bu2Te=O (16) (Scheme 4) [13]. Aldehydes 6 were again used as substrates in reactions to form products 7. No comments were made regarding the relative advantages or disadvantages of using either 3 or 15 as the catalyst for such reactions, and in fact similar alkyl halides 4 were used to generate ylides 16 as were used in the reactions catalyzed by 3. The only notable differences regarding performing the reactions were that reactions with 15 required a higher temperature (50 °C compared to room temperature) and acetonitrile as a co-solvent. However, product yields and stereoselectivities were slightly improved when 15 was used at the same loading level (0.2 equivalents) and with a similar set of aldehyde 6 substrates.
![[1860-5397-12-253-i4]](/bjoc/content/inline/1860-5397-12-253-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Bu2Te-catalyzed Wittig-type reactions.
Scheme 4: Bu2Te-catalyzed Wittig-type reactions.
Tang’s research group also followed up this tellurium-based research many years later and published several papers describing the use of polymer-supported tellurides, such as 18, as catalysts (Scheme 5) [14-16]. The major advantage reported for using 18 instead of 15 is that a much lower catalyst loading could be used in similar reactions (0.02 equivalents compared to 0.2 equivalents). Unfortunately, despite the fact that 19 could be easily reduced to 18 using triphenylphosphite or sodium bisulfite, and simply removed from the desired alkene products, recovered 18 had a much lower activity when attempts to reuse it were made. Nevertheless, the use of 18 with sodium bisulfite as the reducing reagent allowed for very simple product isolation, and reaction yields and stereoselectivities were similar to when 15 was used as the catalyst.
![[1860-5397-12-253-i5]](/bjoc/content/inline/1860-5397-12-253-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Polymer-supported telluride catalyst cycling.
Scheme 5: Polymer-supported telluride catalyst cycling.
In the course of performing the research mentioned above, Tang and co-workers made the fortuitous observation that telluronium salt 20 (prepared from 15) decomposed in the presence of water to form 21. This compound could be used as a pre-catalyst in Wittig-type reactions because it is reduced to 15 by triphenylphosphite in the presence of potassium carbonate (Scheme 6) [17]. Since 21 was observed to be stable and odourless, its use has some practical advantages, and when it was used as a surrogate for 15, very similar results were obtained.
![[1860-5397-12-253-i6]](/bjoc/content/inline/1860-5397-12-253-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Stable and odourless telluronium salt pre-catalyst for Wittig-type reactions.
Scheme 6: Stable and odourless telluronium salt pre-catalyst for Wittig-type reactions.
While these arsenic and tellurium-based reactions are conceptually interesting and show the way in which phosphorous-based catalytic Wittig reactions might be performed, it appears that they have not been used by anyone other than the original reporters.
Phosphine-catalyzed reactions
As alluded to above, it seems that the major impediment to the development of phosphine-catalyzed Wittig reactions was the stability of phosphine oxides and the harsh reaction conditions generally required to reduce them to the corresponding phosphines were thought to be incompatible with various necessary reactants and reagents, and the desired reaction products to be formed.
Christopher J. O’Brien and co-workers recently reported a breakthrough of identifying the necessary selective conditions for phosphine oxide reduction that allowed phosphorous-based catalytic Wittig reactions to become realized. In their initial publication they described the use of readily available phosphine oxide 22 as a pre-catalyst (0.1 equivalents), which was reduced in situ with diphenylsilane (Ph2SiH2) to phosphine 23, which served as the actual catalyst in their Wittig reactions (Scheme 7) [18]. Once 23 was generated, it reacted with electron-withdrawing group activated alkyl halides 4 and sodium carbonate to form the corresponding phosphonium ylides that reacted with aldehydes 6 to produce alkene products 7 and 22 as a byproduct.
![[1860-5397-12-253-i7]](/bjoc/content/inline/1860-5397-12-253-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Phosphine-catalyzed Wittig reactions.
Scheme 7: Phosphine-catalyzed Wittig reactions.
Subsequently they reported that the soluble organic base N,N-diisopropylethylamine was a good replacement for sodium carbonate in such reactions [19], and that the addition of 4-nitrobenzoic acid facilitated the phosphine oxide reduction step using phenylsilane (PhSiH3) instead of diphenylsilane [20]. Using this combination of 4-nitrobenzoic acid and phenylsilane for phosphine oxide reduction allowed reactions starting with phosphine oxide 24 (Figure 2) to be conducted at room temperature and for acyclic phosphine oxides 2 and 25 to be used as well, albeit at elevated operating temperature. Most recently they have found that the use of 26 as the pre-catalyst in conjunction with sodium tert-butoxycarbonate (NaOCO2t-Bu, a slow release form of sodium tert-butoxide) as a precursor for the required base, catalytic Wittig reactions could be performed using semi or non-stabilized ylides [21].
![[1860-5397-12-253-2]](/bjoc/content/figures/1860-5397-12-253-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Various phosphine oxides used as pre-catalysts.
Figure 2: Various phosphine oxides used as pre-catalysts.
Thomas Werner’s research group has also been active in this area of research, and reported the first example of a catalytic enantioselective Wittig reaction (Scheme 8) [22]. This reaction involved the intramolecular cyclization of 27 to form 28. A variety of phosphines were examined as the catalyst, and (S,S)-29 ((S,S)-Me-DuPhos, 0.1 equivalent) was found to provide the best combination of reactivity and stereoselectivity (39% yield, 62% ee). In these reactions butylene oxide was used as a base precursor, phenylsilane was the reducing reagent, and the reactions were performed using microwave irradiation (MWI). Subsequently, Werner et al. reported the scope and limitations of such microwave-assisted catalytic Wittig reactions using tributylphosphine, phenylsilane and butylene oxide [23,24], and reactions with the combination of achiral 30, trimethoxysilane ((MeO)3SiH) and sodium carbonate using conventional heating [25].
![[1860-5397-12-253-i8]](/bjoc/content/inline/1860-5397-12-253-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Enantioselective catalytic Wittig reaction reported by Werner.
Scheme 8: Enantioselective catalytic Wittig reaction reported by Werner.
Additionally, the same authors have reported base-free Wittig reactions using diethyl maleate (31) as the starting material to form products 32 catalyzed by tributylphosphine (33, 0.05 equivalents) (Scheme 9) [26]. In these transformations the initial reaction between 31 and 33 generated zwitterion 34, that underwent internal proton transfer to generate ylide 35. This in turn reacted with aldehyde 6 to form 32 and phenylsilane was used as the reducing reagent to regenerate 33. Most recently they refined such reactions using 22 as the pre-catalyst, trimethoxysilane as the reducing reagent, and catalytic benzoic acid to facilitate phosphine oxide reduction [27].
![[1860-5397-12-253-i9]](/bjoc/content/inline/1860-5397-12-253-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Base-free catalytic Wittig reactions reported by Werner.
Scheme 9: Base-free catalytic Wittig reactions reported by Werner.
At about the same time as the penultimate report by Werner and co-workers appeared, Wenwei Lin and a co-worker published conceptually similar catalytic Wittig reactions that were based on their previous research regarding related non-catalytic phosphine-mediated base-free Wittig reactions (Scheme 10) [28]. They started with Michael acceptors 36 to generate products 37 using 22 as the pre-catalyst with triethylamine as the base, phenylsilane as the reducing reagent, and 4-nitrobenzoic acid as an acidic additive. It should be noted that the role of the base in these reactions is unclear and not directly commented on. According to the proposed mechanism for the formation of the required ylide intermediate, a base is not necessary, but the authors reported that when it was omitted from a control reaction, no reaction occurred.
![[1860-5397-12-253-i10]](/bjoc/content/inline/1860-5397-12-253-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Catalytic Wittig reactions reported by Lin.
Scheme 10: Catalytic Wittig reactions reported by Lin.
Finally, Bernd Plietker and co-workers have very recently reported the use of iron complex 38 as a catalyst for phosphine oxide reduction and have incorporated it into Wittig reactions catalyzed by 1 (Scheme 11) [29]. These reactions are similar to those mentioned previously by O’Brien’s group [20] that involve the cycling between 1 and 2 using a silane reducing reagent.
![[1860-5397-12-253-i11]](/bjoc/content/inline/1860-5397-12-253-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Catalytic Wittig reactions reported by Plietker.
Scheme 11: Catalytic Wittig reactions reported by Plietker.
As can be seen above, the issue of selective phosphine oxide reduction has been solved using various silane reagents and much progress has been made in phosphine-catalyzed Wittig reactions. Initial results were reported using phosphine oxides that were prepared from commercially available phosphine oxide starting materials that were relatively easy to reduce, such as 22. However, relatively mild cycling between 1 and 2 can now be achieved, and this may make such catalytic Wittig reactions more popular, practical, and scalable due to the stability, wide availability and low cost of 1.
Catalytic aza-Wittig reactions
Aza-Wittig reactions are similar to Wittig reactions in that they also involve the reaction of a phosphonium ylide, in this case an iminophosphorane (or phosphinimide) such as 39, with a carbonyl group containing compound to form the carbon–nitrogen double bond of an imine along with a byproduct phosphine oxide such as 2 (Scheme 12). The difference is that the iminophosphorane reagents can be generated either from a phosphine such as 1 or from a phosphine oxide such as 2, by reaction with either an azide or isocyanate reagent, respectively. Thus, two possible strategies for catalytic aza-Wittig reactions exist, one using a phosphorous(V) catalyst, and the other using a phosphorous(III) catalyst that is regenerated in the catalytic cycle. These strategies are the topic of this section of the review.
![[1860-5397-12-253-i12]](/bjoc/content/inline/1860-5397-12-253-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Prototypical aza-Wittig reaction involving in situ iminophosphorane formation.
Scheme 12: Prototypical aza-Wittig reaction involving in situ iminophosphorane formation.
Phosphine oxide-catalyzed reactions
It is clear from Scheme 12 that when a phosphine oxide is used to generate the iminophosphorane 39 for an aza-Wittig reaction, it is regenerated as a byproduct, and thus can participate directly in another reaction cycle. More than 25 years before Huang’s research group made their first report regarding arsine catalysis [10], Tod W. Campbell and colleagues took advantage of this fact and reported a single example of a catalytic aza-Wittig reaction as part of their research on phosphine oxide-catalyzed carbodiimide synthesis (Scheme 13) [30]. In their reaction they used phosphine oxide 40 as the catalyst in the reaction between diisocyanate 41 and benzaldehyde (42, 2 equivalents) to form diimine 43 and carbon dioxide (2 equivalents). While this reaction was described as being catalytic, the mass of the catalyst used was not explicitly reported, so it is impossible to determine the number of catalyst turnovers that were involved in generating the 20% isolated yield of 43. The authors only reported that “one drop” of 40 was used in a reaction starting with 7 grams of 41.
![[1860-5397-12-253-i13]](/bjoc/content/inline/1860-5397-12-253-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: First catalytic aza-Wittig reaction reported by Campbell.
Scheme 13: First catalytic aza-Wittig reaction reported by Campbell.
Subsequently, Stephen P. Marsden and co-workers reported intramolecular versions of catalytic aza-Wittig reactions for heteroaromatic compound synthesis using commercially available phosphine oxide 44 as the catalyst (Scheme 14) [31]. In this work, biaryl isocyanates 45 could be converted into phenanthridines 46, and aryl isocyanates 47 could be transformed into benzoxazoles 48 directly in refluxing toluene together with the simultaneous release of carbon dioxide. Presumably these reactions proceeded via iminophosphorane intermediates that reacted intramolecularly with the carbonyl groups to form the obtained cyclic products. It is noteworthy that the loading of catalyst 44 could be as low as 0.01 equivalent.
![[1860-5397-12-253-i14]](/bjoc/content/inline/1860-5397-12-253-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Intramolecular catalytic aza-Wittig reactions reported by Marsden.
Scheme 14: Intramolecular catalytic aza-Wittig reactions reported by Marsden.
More recently the research group of Ming-Wu Ding has extended this concept of phosphine oxide-catalyzed aza-Wittig reactions to the conversion of carboxylic acid derivatives 49 into 1,4-benzodiazepin-5-ones 50 using catalyst 44 (Scheme 15) [32]. The overall transformations were reported to occur via acyl azide intermediates 51 that were not purified, but instead used directly in thermal Curtius rearrangement reactions that afforded isocyanates 52. These were in turn treated in situ with catalyst 44 to afford the final products 50 via presumed iminophosphorane intermediates 53.
![[1860-5397-12-253-i15]](/bjoc/content/inline/1860-5397-12-253-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Catalytic aza-Wittig reactions in 1,4-benzodiazepin-5-one synthesis.
Scheme 15: Catalytic aza-Wittig reactions in 1,4-benzodiazepin-5-one synthesis.
Subsequently this research group used a very similar strategy for the synthesis of polysubstituted benzimidazoles 54 via sequential Ugi and catalytic aza-Wittig reactions (Scheme 16) [33]. It was reported that mixing 2-aminobenzoyl azides 55, carboxylic acids 56, isocyanides 57 with aldehydes 6 in methanol generated intermediates 58, which underwent rearrangement to isocyanates 59 in refluxing toluene. Finally, catalytic aza-Wittig reactions with 44 produced cyclized final products 54 via iminophosphoranes 60.
![[1860-5397-12-253-i16]](/bjoc/content/inline/1860-5397-12-253-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Catalytic aza-Wittig reactions in benzimidazole synthesis.
Scheme 16: Catalytic aza-Wittig reactions in benzimidazole synthesis.
Phosphine-catalyzed reactions
The initial research in this area was performed by Floris L. van Delft and co-workers who reported the synthesis of 61 and its use in catalytic Staudinger reactions for the reduction of azides 62 to amines 63 (Scheme 17) [34]. Subsequently they extended such reactions to include aza-Wittig reactions using diphenylsilane as the reducing reagent [35]. For example, starting materials 64 could be converted into benzoxazoles 48 in overall net transformations that were similar as to those discussed above by Marsden (Scheme 14). In their report they also described the synthesis of various other classes of heterocyclic compounds such as 3H-1,4-benzodiazepin-2(1H)-ones 65, 3H-1,4-benzodiazepin-5(4H)-ones 66, and pyrrole 67 from the corresponding starting materials using similar reaction conditions.
![[1860-5397-12-253-i17]](/bjoc/content/inline/1860-5397-12-253-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Phosphine-catalyzed Staudinger and aza-Wittig reactions.
Scheme 17: Phosphine-catalyzed Staudinger and aza-Wittig reactions.
In addition to phosphine oxide-catalyzed aza-Wittig reactions, Ding’s research group has also explored the use of phosphine catalysis in such reactions. In their initial report regarding this strategy, they used 1 to catalyze intramolecular reactions that converted aryl azides 68 into 4(3H)-quinazolinones 69 via intermediate iminophosphoranes 70, using the combination of titanium tetraisopropoxide and tetramethyldisiloxane (TMDS) for the in situ reduction of byproduct 2 (Scheme 18) [36].
![[1860-5397-12-253-i18]](/bjoc/content/inline/1860-5397-12-253-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Catalytic aza-Wittig reactions in 4(3H)-quinazolinone synthesis.
Scheme 18: Catalytic aza-Wittig reactions in 4(3H)-quinazolinone synthesis.
This group has more recently studied catalytic aza-Wittig reactions using carboxylic acid anhydrides as the starting materials (Scheme 19) [37]. For example, reactions of carboxylic acids 71 with acid chlorides 72 to generate the corresponding carboxylic acid anhydride in situ afforded 4H-benzo[d][1,3]-oxazin-4-ones 73. In these reactions, 1 was used as the catalyst for the aza-Wittig reaction and copper triflate was used as the catalyst for phosphine oxide reduction. Using similar conditions the corresponding reactions of carboxylic acids 74 with aromatic acid chlorides 72 produced 4-benzylidene-2-aryloxazol-5(4H)-ones 75.
![[1860-5397-12-253-i19]](/bjoc/content/inline/1860-5397-12-253-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Catalytic aza-Wittig reactions of in situ generated carboxylic acid anhydrides.
Scheme 19: Catalytic aza-Wittig reactions of in situ generated carboxylic acid anhydrides.
Lastly, the research group of Piet Herdewijn extended this general concept and reported catalytic diaza-Wittig reactions (Scheme 20) [38]. In these reactions 76 (from the reduction of 44) was the catalyst that transformed diazo group containing starting materials 77 into pyridazine derivatives 78. In these reactions 44 was actually added to the reaction mixtures as the pre-catalyst that was reduced using diphenylsilane.
![[1860-5397-12-253-i20]](/bjoc/content/inline/1860-5397-12-253-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Phosphine-catalyzed diaza-Wittig reactions.
Scheme 20: Phosphine-catalyzed diaza-Wittig reactions.
It is readily evident from the above examples that regardless of whether phosphine or phosphine oxides were used as the catalyst, catalytic aza-Wittig reactions have emerged to become powerful tools in the synthesis of collections of various heterocyclic compounds since it seems that the required isocyanate or azide group containing precursors are readily synthesized from simple starting materials.
Conclusion
While great advances have been reported regarding the development of catalytic Wittig and aza-Wittig reactions, it remains to be seen how widely these methods will be adopted. Evidence that the former reactions are being described in the literature with increasing frequency are the very recent reports by Saleh and Voituriez [39], and Wenwei Lin and co-workers [40] regarding intramolecular reactions to form heterocycles that appeared as this review was being completed. However, will the research results summarized herein remain merely intellectual achievements or will they become commonly used synthetic methods in the future? Perhaps the answer to this question is how these new reaction systems are viewed from an environmental/green chemistry perspective. In this regard it is encouraging that a life cycle assessment indicates that the use of stoichiometric quantities of silanes as replacements for phosphines in catalytic Wittig reactions can offer environmental improvements [41]. Thus it seems somewhat reasonable to expect that as even more efficient methods for phosphine oxide reduction are discovered [42-47], catalytic reactions involving cycling between phosphines and phosphine oxides will become more environmentally friendly and more popular too. With regards to the phosphine oxide-catalyzed aza-Wittig reactions discussed, while no life cycle assessments have been performed, they do seem to be rather “green” since no redox cycling is necessary, and there appear to be few constraints to their potential application.
References
-
Wittig, G.; Geissler, G. Justus Liebigs Ann. Chem. 1953, 580, 44–57. doi:10.1002/jlac.19535800107
Return to citation in text: [1] -
Byrne, P. A.; Gilheany, D. G. Chem. Soc. Rev. 2013, 42, 6670–6696. doi:10.1039/C3CS60105F
Return to citation in text: [1] -
Xu, S.; He, Z. RSC Adv. 2013, 3, 16885–16904. doi:10.1039/C3RA42088D
Return to citation in text: [1] -
McNulty, J.; McLeod, D.; Das, P.; Zepeda-Velázquez, C. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 619–632. doi:10.1080/10426507.2014.980907
Return to citation in text: [1] -
Fairlamb, I. J. S. ChemSusChem 2009, 2, 1021–1024. doi:10.1002/cssc.200900208
Return to citation in text: [1] -
van Kalkeren, H. A.; van Delft, F. L.; Rutjes, F. P. J. T. ChemSusChem 2013, 6, 1615–1624. doi:10.1002/cssc.201300368
Return to citation in text: [1] -
Xu, S.; Tang, Y. Lett. Org. Chem. 2014, 11, 524–533. doi:10.2174/1570178611666140401223253
Return to citation in text: [1] -
Voituriez, A.; Saleh, N. Tetrahedron Lett. 2016, 57, 4443–4451. doi:10.1016/j.tetlet.2016.08.036
Return to citation in text: [1] -
He, H. S.; Chung, C. W. Y.; But, T. Y. S.; Toy, P. H. Tetrahedron 2005, 61, 1385–1405. doi:10.1016/j.tet.2004.11.031
Return to citation in text: [1] -
Shi, L.; Wang, W.; Wang, Y.; Huang, Y. J. Org. Chem. 1989, 54, 2027–2028. doi:10.1021/jo00270a001
Return to citation in text: [1] [2] -
Cao, P.; Li, C.-Y.; Kang, Y.-B.; Xie, Z.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2007, 72, 6628–6630. doi:10.1021/jo0709899
Return to citation in text: [1] -
Wang, P.; Liu, C.-R.; Sun, X.-L.; Chen, S.-S.; Li, J.-F.; Xie, Z.; Tang, Y. Chem. Commun. 2012, 48, 290–292. doi:10.1039/C1CC16747B
Return to citation in text: [1] -
Huang, Y.-Z.; Shi, L.-L.; Li, S.-W.; Wen, X.-Q. J. Chem. Soc., Perkin Trans. 1 1989, 2397–2399. doi:10.1039/P19890002397
Return to citation in text: [1] -
Huang, Z.-Z.; Ye, S.; Xia, W.; Tang, Y. Chem. Commun. 2001, 1384–1385. doi:10.1039/B104100M
Return to citation in text: [1] -
Huang, Z.-Z.; Ye, S.; Xia, W.; Yu, Y.-H.; Tang, Y. J. Org. Chem. 2002, 67, 3096–3103. doi:10.1021/jo025586h
Return to citation in text: [1] -
Li, K.; Ran, L.; Yu, Y.-H.; Tang, Y. J. Org. Chem. 2004, 69, 3986–3989. doi:10.1021/jo049701v
Return to citation in text: [1] -
Huang, Z.-Z.; Tang, Y. J. Org. Chem. 2002, 67, 5320–5326. doi:10.1021/jo025693b
Return to citation in text: [1] -
O’Brien, C. J.; Tellez, J. L.; Nixon, Z. S.; Kang, L. J.; Carter, A. L.; Kunkel, S. R.; Przeworski, K. C.; Chass, G. A. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. doi:10.1002/anie.200902525
Return to citation in text: [1] -
O’Brien, C. J.; Nixon, Z. S.; Holohan, A. J.; Kunkel, S. R.; Tellez, J. L.; Doonan, B. J.; Coyle, E. J.; Lavigne, F.; Kang, L. J.; Przeworski, K. C. Chem. – Eur. J. 2013, 19, 15281–15289. doi:10.1002/chem.201301444
Return to citation in text: [1] -
O’Brien, C. J.; Lavigne, F.; Coyle, E. E.; Holohan, A. J.; Doonan, B. J. Chem. – Eur. J. 2013, 19, 5854–5858. doi:10.1002/chem.201300546
Return to citation in text: [1] [2] -
Coyle, E. E.; Doonan, B. J.; Holohan, A. J.; Walsh, K. A.; Lavigne, F.; Krenske, E. H.; O’Brien, C. J. Angew. Chem., Int. Ed. 2014, 53, 12907–12911. doi:10.1002/anie.201406103
Return to citation in text: [1] -
Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2014, 6630–6633. doi:10.1002/ejoc.201402941
Return to citation in text: [1] -
Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2014, 6873–6876. doi:10.1002/ejoc.201403113
Return to citation in text: [1] -
Hoffmann, M.; Deshmukh, S.; Werner, T. Eur. J. Org. Chem. 2015, 4532–4543. doi:10.1002/ejoc.201500310
Return to citation in text: [1] -
Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2015, 3286–3295. doi:10.1002/ejoc.201500243
Return to citation in text: [1] -
Schirmer, M.-L.; Adomeit, S.; Werner, T. Org. Lett. 2015, 17, 3078–3081. doi:10.1021/acs.orglett.5b01352
Return to citation in text: [1] -
Schirmer, M.-L.; Adomelt, S.; Spannenberg, A.; Werner, T. Chem. – Eur. J. 2016, 22, 2458–2465. doi:10.1002/chem.201503744
Return to citation in text: [1] -
Tsai, Y.-L.; Lin, W. Asian J. Org. Chem. 2015, 4, 1040–1043. doi:10.1002/ajoc.201500251
Return to citation in text: [1] -
Rommel, S.; Belger, C.; Begouin, J.-M.; Plietker, B. ChemCatChem 2015, 7, 1292–1301. doi:10.1002/cctc.201500053
Return to citation in text: [1] -
Campbell, T. W.; Monagle, J. J.; Foldi, V. S. J. Am. Chem. Soc. 1962, 84, 3673–3677. doi:10.1021/ja00878a015
Return to citation in text: [1] -
Marsden, S. P.; McGonagle, A. E.; McKeever-Abbas, B. Org. Lett. 2008, 10, 2589–2591. doi:10.1021/ol800921n
Return to citation in text: [1] -
Wang, L.; Qin, R.-Q.; Yan, H.-Y.; Ding, M.-W. Synthesis 2015, 47, 3522–3528. doi:10.1055/s-0034-1378874
Return to citation in text: [1] -
Yan, Y.-M.; Rao, Y.; Ding, M.-W. J. Org. Chem. 2016, 81, 1263–1268. doi:10.1021/acs.joc.5b02575
Return to citation in text: [1] -
van Kalkeren, H. A.; Bruins, J. J.; Rutjes, F. P. J. T.; van Delft, F. L. Adv. Synth. Catal. 2012, 354, 1417–1421. doi:10.1002/adsc.201100967
Return to citation in text: [1] -
van Kalkeren, H. A.; te Grotenhuis, C.; Haasjes, F. S.; Hommerson, C. R. A.; Rutjes, F. P. J. T.; van Delft, F. L. Eur. J. Org. Chem. 2013, 7059–7066. doi:10.1002/ejoc.201300585
Return to citation in text: [1] -
Wang, L.; Wang, Y.; Chen, M.; Ding, M.-W. Adv. Synth. Catal. 2014, 356, 1098–1104. doi:10.1002/adsc.201300950
Return to citation in text: [1] -
Wang, L.; Xie, Y.-B.; Huang, N.-Y.; Yan, J.-Y.; Hu, W.-M.; Liu, M.-G.; Ding, M.-W. ACS Catal. 2016, 6, 4010–4016. doi:10.1021/acscatal.6b00165
Return to citation in text: [1] -
Bel Abed, H.; Mammoliti, O.; Bande, O.; Van Lommen, G.; Herdewijn, P. Org. Biomol. Chem. 2014, 12, 7159–7166. doi:10.1039/C4OB01201A
Return to citation in text: [1] -
Saleh, N.; Voituriez, A. J. Org. Chem. 2016, 81, 4371–4377. doi:10.1021/acs.joc.6b00473
Return to citation in text: [1] -
Lee, C.-J.; Chang, T.-H.; Yu, J.-K.; Reddy, G. M.; Hsiao, M.-Y.; Lin, W. Org. Lett. 2016, 18, 3758–3761. doi:10.1021/acs.orglett.6b01781
Return to citation in text: [1] -
van Kalkeren, H. A.; Blom, A. L.; Rutjes, F. P. J. T.; Huijbregts, M. A. J. Green Chem. 2013, 15, 1255–1263. doi:10.1039/C3GC00053B
Return to citation in text: [1] -
Kuroboshi, M.; Yano, T.; Kamenoue, S.; Kawakubo, H.; Tanaka, H. Tetrahedron 2011, 67, 5825–5831. doi:10.1016/j.tet.2011.05.044
Return to citation in text: [1] -
Li, Y.; Lu, L.-Q.; Das, S.; Pisiewicz, S.; Junge, K.; Beller, M. J. Am. Chem. Soc. 2012, 134, 18325–18329. doi:10.1021/ja3069165
Return to citation in text: [1] -
Keglevich, G.; Kovács, T.; Csatlós, F. Heteroat. Chem. 2015, 26, 199–205. doi:10.1002/hc.21249
Return to citation in text: [1] -
Demchuk, O. M.; Jasiński, R.; Pietrusiewicz, K. M. Heteroat. Chem. 2015, 26, 441–448. doi:10.1002/hc.21279
Return to citation in text: [1] -
Schirmer, M.-L.; Jopp, S.; Holz, J.; Spannenberg, A.; Werner, T. Adv. Synth. Catal. 2016, 358, 26–29. doi:10.1002/adsc.201500762
Return to citation in text: [1] -
Hérault, D.; Nguyen, D. H.; Nuel, D.; Buono, G. Chem. Soc. Rev. 2015, 44, 2508–2528. doi:10.1039/C4CS00311J
Return to citation in text: [1]
| 35. | van Kalkeren, H. A.; te Grotenhuis, C.; Haasjes, F. S.; Hommerson, C. R. A.; Rutjes, F. P. J. T.; van Delft, F. L. Eur. J. Org. Chem. 2013, 7059–7066. doi:10.1002/ejoc.201300585 |
| 36. | Wang, L.; Wang, Y.; Chen, M.; Ding, M.-W. Adv. Synth. Catal. 2014, 356, 1098–1104. doi:10.1002/adsc.201300950 |
| 37. | Wang, L.; Xie, Y.-B.; Huang, N.-Y.; Yan, J.-Y.; Hu, W.-M.; Liu, M.-G.; Ding, M.-W. ACS Catal. 2016, 6, 4010–4016. doi:10.1021/acscatal.6b00165 |
| 1. | Wittig, G.; Geissler, G. Justus Liebigs Ann. Chem. 1953, 580, 44–57. doi:10.1002/jlac.19535800107 |
| 2. | Byrne, P. A.; Gilheany, D. G. Chem. Soc. Rev. 2013, 42, 6670–6696. doi:10.1039/C3CS60105F |
| 11. | Cao, P.; Li, C.-Y.; Kang, Y.-B.; Xie, Z.; Sun, X.-L.; Tang, Y. J. Org. Chem. 2007, 72, 6628–6630. doi:10.1021/jo0709899 |
| 23. | Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2014, 6873–6876. doi:10.1002/ejoc.201403113 |
| 24. | Hoffmann, M.; Deshmukh, S.; Werner, T. Eur. J. Org. Chem. 2015, 4532–4543. doi:10.1002/ejoc.201500310 |
| 9. | He, H. S.; Chung, C. W. Y.; But, T. Y. S.; Toy, P. H. Tetrahedron 2005, 61, 1385–1405. doi:10.1016/j.tet.2004.11.031 |
| 10. | Shi, L.; Wang, W.; Wang, Y.; Huang, Y. J. Org. Chem. 1989, 54, 2027–2028. doi:10.1021/jo00270a001 |
| 25. | Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2015, 3286–3295. doi:10.1002/ejoc.201500243 |
| 5. | Fairlamb, I. J. S. ChemSusChem 2009, 2, 1021–1024. doi:10.1002/cssc.200900208 |
| 6. | van Kalkeren, H. A.; van Delft, F. L.; Rutjes, F. P. J. T. ChemSusChem 2013, 6, 1615–1624. doi:10.1002/cssc.201300368 |
| 7. | Xu, S.; Tang, Y. Lett. Org. Chem. 2014, 11, 524–533. doi:10.2174/1570178611666140401223253 |
| 8. | Voituriez, A.; Saleh, N. Tetrahedron Lett. 2016, 57, 4443–4451. doi:10.1016/j.tetlet.2016.08.036 |
| 21. | Coyle, E. E.; Doonan, B. J.; Holohan, A. J.; Walsh, K. A.; Lavigne, F.; Krenske, E. H.; O’Brien, C. J. Angew. Chem., Int. Ed. 2014, 53, 12907–12911. doi:10.1002/anie.201406103 |
| 42. | Kuroboshi, M.; Yano, T.; Kamenoue, S.; Kawakubo, H.; Tanaka, H. Tetrahedron 2011, 67, 5825–5831. doi:10.1016/j.tet.2011.05.044 |
| 43. | Li, Y.; Lu, L.-Q.; Das, S.; Pisiewicz, S.; Junge, K.; Beller, M. J. Am. Chem. Soc. 2012, 134, 18325–18329. doi:10.1021/ja3069165 |
| 44. | Keglevich, G.; Kovács, T.; Csatlós, F. Heteroat. Chem. 2015, 26, 199–205. doi:10.1002/hc.21249 |
| 45. | Demchuk, O. M.; Jasiński, R.; Pietrusiewicz, K. M. Heteroat. Chem. 2015, 26, 441–448. doi:10.1002/hc.21279 |
| 46. | Schirmer, M.-L.; Jopp, S.; Holz, J.; Spannenberg, A.; Werner, T. Adv. Synth. Catal. 2016, 358, 26–29. doi:10.1002/adsc.201500762 |
| 47. | Hérault, D.; Nguyen, D. H.; Nuel, D.; Buono, G. Chem. Soc. Rev. 2015, 44, 2508–2528. doi:10.1039/C4CS00311J |
| 3. | Xu, S.; He, Z. RSC Adv. 2013, 3, 16885–16904. doi:10.1039/C3RA42088D |
| 4. | McNulty, J.; McLeod, D.; Das, P.; Zepeda-Velázquez, C. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 619–632. doi:10.1080/10426507.2014.980907 |
| 22. | Werner, T.; Hoffmann, M.; Deshmukh, S. Eur. J. Org. Chem. 2014, 6630–6633. doi:10.1002/ejoc.201402941 |
| 17. | Huang, Z.-Z.; Tang, Y. J. Org. Chem. 2002, 67, 5320–5326. doi:10.1021/jo025693b |
| 19. | O’Brien, C. J.; Nixon, Z. S.; Holohan, A. J.; Kunkel, S. R.; Tellez, J. L.; Doonan, B. J.; Coyle, E. J.; Lavigne, F.; Kang, L. J.; Przeworski, K. C. Chem. – Eur. J. 2013, 19, 15281–15289. doi:10.1002/chem.201301444 |
| 40. | Lee, C.-J.; Chang, T.-H.; Yu, J.-K.; Reddy, G. M.; Hsiao, M.-Y.; Lin, W. Org. Lett. 2016, 18, 3758–3761. doi:10.1021/acs.orglett.6b01781 |
| 14. | Huang, Z.-Z.; Ye, S.; Xia, W.; Tang, Y. Chem. Commun. 2001, 1384–1385. doi:10.1039/B104100M |
| 15. | Huang, Z.-Z.; Ye, S.; Xia, W.; Yu, Y.-H.; Tang, Y. J. Org. Chem. 2002, 67, 3096–3103. doi:10.1021/jo025586h |
| 16. | Li, K.; Ran, L.; Yu, Y.-H.; Tang, Y. J. Org. Chem. 2004, 69, 3986–3989. doi:10.1021/jo049701v |
| 20. | O’Brien, C. J.; Lavigne, F.; Coyle, E. E.; Holohan, A. J.; Doonan, B. J. Chem. – Eur. J. 2013, 19, 5854–5858. doi:10.1002/chem.201300546 |
| 41. | van Kalkeren, H. A.; Blom, A. L.; Rutjes, F. P. J. T.; Huijbregts, M. A. J. Green Chem. 2013, 15, 1255–1263. doi:10.1039/C3GC00053B |
| 13. | Huang, Y.-Z.; Shi, L.-L.; Li, S.-W.; Wen, X.-Q. J. Chem. Soc., Perkin Trans. 1 1989, 2397–2399. doi:10.1039/P19890002397 |
| 38. | Bel Abed, H.; Mammoliti, O.; Bande, O.; Van Lommen, G.; Herdewijn, P. Org. Biomol. Chem. 2014, 12, 7159–7166. doi:10.1039/C4OB01201A |
| 12. | Wang, P.; Liu, C.-R.; Sun, X.-L.; Chen, S.-S.; Li, J.-F.; Xie, Z.; Tang, Y. Chem. Commun. 2012, 48, 290–292. doi:10.1039/C1CC16747B |
| 18. | O’Brien, C. J.; Tellez, J. L.; Nixon, Z. S.; Kang, L. J.; Carter, A. L.; Kunkel, S. R.; Przeworski, K. C.; Chass, G. A. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. doi:10.1002/anie.200902525 |
| 39. | Saleh, N.; Voituriez, A. J. Org. Chem. 2016, 81, 4371–4377. doi:10.1021/acs.joc.6b00473 |
| 28. | Tsai, Y.-L.; Lin, W. Asian J. Org. Chem. 2015, 4, 1040–1043. doi:10.1002/ajoc.201500251 |
| 26. | Schirmer, M.-L.; Adomeit, S.; Werner, T. Org. Lett. 2015, 17, 3078–3081. doi:10.1021/acs.orglett.5b01352 |
| 27. | Schirmer, M.-L.; Adomelt, S.; Spannenberg, A.; Werner, T. Chem. – Eur. J. 2016, 22, 2458–2465. doi:10.1002/chem.201503744 |
| 33. | Yan, Y.-M.; Rao, Y.; Ding, M.-W. J. Org. Chem. 2016, 81, 1263–1268. doi:10.1021/acs.joc.5b02575 |
| 34. | van Kalkeren, H. A.; Bruins, J. J.; Rutjes, F. P. J. T.; van Delft, F. L. Adv. Synth. Catal. 2012, 354, 1417–1421. doi:10.1002/adsc.201100967 |
| 31. | Marsden, S. P.; McGonagle, A. E.; McKeever-Abbas, B. Org. Lett. 2008, 10, 2589–2591. doi:10.1021/ol800921n |
| 32. | Wang, L.; Qin, R.-Q.; Yan, H.-Y.; Ding, M.-W. Synthesis 2015, 47, 3522–3528. doi:10.1055/s-0034-1378874 |
| 10. | Shi, L.; Wang, W.; Wang, Y.; Huang, Y. J. Org. Chem. 1989, 54, 2027–2028. doi:10.1021/jo00270a001 |
| 30. | Campbell, T. W.; Monagle, J. J.; Foldi, V. S. J. Am. Chem. Soc. 1962, 84, 3673–3677. doi:10.1021/ja00878a015 |
| 29. | Rommel, S.; Belger, C.; Begouin, J.-M.; Plietker, B. ChemCatChem 2015, 7, 1292–1301. doi:10.1002/cctc.201500053 |
| 20. | O’Brien, C. J.; Lavigne, F.; Coyle, E. E.; Holohan, A. J.; Doonan, B. J. Chem. – Eur. J. 2013, 19, 5854–5858. doi:10.1002/chem.201300546 |
© 2016 Lao and Toy; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)