Abstract
We describe the synthesis of bromo-tert-butyloxycarbonyl (Br-t-BOC)-amino-protected monomers 2-((1-bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl (meth)acrylate 3a,b. For this purpose, 2-isocyanatoethyl (meth)acrylate 1a,b was reacted with 1-bromo-2-methylpropan-2-ol (2a). The free radical polymerization of (Br-t-BOC)-aminoethyl (meth)acrylates 3a,b yielded poly((Br-t-BOC)-aminoethyl (meth)acrylate) 6a,b bearing protected amino side groups. The subsequent solvolysis of the Br-t-BOC function led to the new polymers poly(2-aminoethyl (meth)acrylate) 8a,b with protonated free amino groups. The monomers and the resulting polymers were thoroughly characterized by 1H NMR, IR, GPC and DSC methods. The kinetics of the deprotection step was followed by 1H NMR spectroscopy. The solvent polarity and neighboring group effects on the kinetics of deprotection are discussed.

Graphical Abstract
Introduction
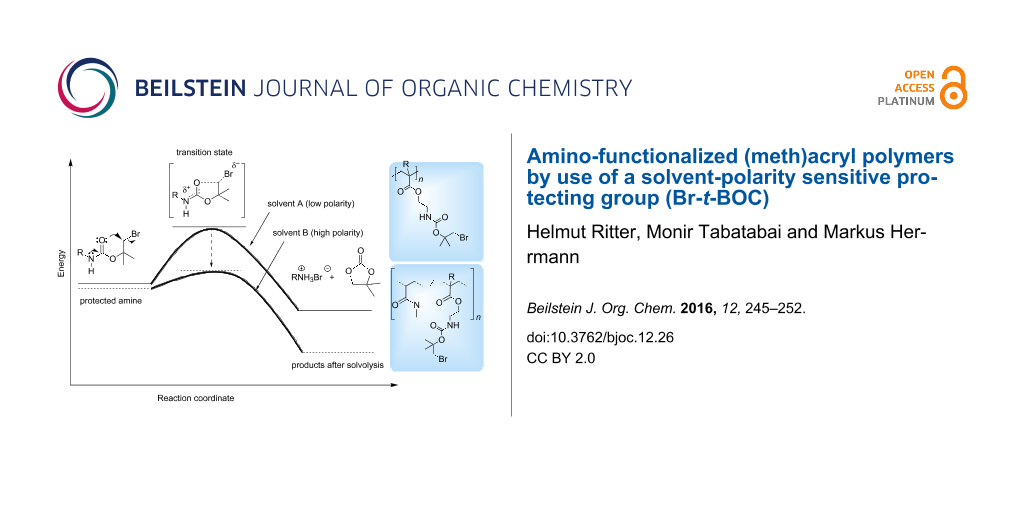
Amino groups are important functionalities in polymer chemistry, e.g., for hardening various epoxy resins [1]. However, they easily react in an undesired side reaction with electron-poor double bonds of (meth)acrylates [2]. Therefore, for the synthesis of amino-containing (meth)acrylic monomers and polymers suitable amino-protecting groups are required. Classical protecting groups such as ammonium salts, F-MOC, Z- or t-BOC, respectively are readily available. Regarding to this aspect, we published some papers about polymer protecting groups about three decades ago [3-7]. However, they are limited in application by certain restrictions on the deprotection conditions. Therefore, there is a continued interest in developing new protecting groups which can be cleaved by different mechanisms. Keeping this in mind, the bromo-tert-butyloxycarbonyl (Br-t-BOC) group represents the first known solvent-polarity sensitive amino-protecting group. As shown in Figure 1, this group is stable in nonpolar solvents because of high activation energy and easily decomposes in a more polar environment because of reduced activation energy. This effect is a result of an increased polarity of the transition state in comparison to the starting molecule (Figure 1). In contrast to the classical t-BOC protecting group, the Br-t-BOC group can be easily removed without pH adjustments. Since the published papers from L. A. Carpino [8-10] and a first practical application in peptide synthesis [11] this protecting group was quasi forgotten.
![[1860-5397-12-26-1]](/bjoc/content/figures/1860-5397-12-26-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic representation of a deprotection taking the relatively high polarity of the transition state and the solvent polarity into account: high activation energy in nonpolar solvents and low activation energy in polar solvents.
Figure 1: Schematic representation of a deprotection taking the relatively high polarity of the transition st...
In the present work we report new Br-t-Boc-protected (meth)acrylic monomers and their polymerization through free radical polymerization. The kinetics of Br-t-BOC solvolysis of the monomers and polymers in a polar solvent are described.
Results and Discussion
The Br-t-Boc-protected monomers 2-((1-bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl acrylate (3a) and 2-((1-bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl methacrylate (3b) were synthesized by the reaction of 1-bromo-2-methylpropan-2-ol (2a) with 2-isocyanatoethyl (meth)acrylate (1a,b) (Scheme 1). The success of the reaction can be easily shown by, e.g., the disappearance of the N=C=O peak of 1a at 2260 cm−1 and the appearance of an N–H peak at 3350 cm−1 in the IR spectra. Pure samples of 3a,b could be isolated by column chromatography and the 1H NMR spectra of 3a and 3b are shown in Figure 2 and Figure 3.
![[1860-5397-12-26-i1]](/bjoc/content/inline/1860-5397-12-26-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of Br-t-Boc-protected monomers 3a,b and homopolymers 6a,b.
Scheme 1: Synthesis of Br-t-Boc-protected monomers 3a,b and homopolymers 6a,b.
![[1860-5397-12-26-2]](/bjoc/content/figures/1860-5397-12-26-2.png?scale=2.56&max-width=1024&background=FFFFFF)
Figure 2: 300 MHz 1H NMR spectrum of 3a in CDCl3.
Figure 2: 300 MHz 1H NMR spectrum of 3a in CDCl3.
![[1860-5397-12-26-3]](/bjoc/content/figures/1860-5397-12-26-3.png?scale=2.6&max-width=1024&background=FFFFFF)
Figure 3: 300 MHz 1H NMR spectrum of 3b in CDCl3.
Figure 3: 300 MHz 1H NMR spectrum of 3b in CDCl3.
After dissolving monomers 3a and 3b in a polar solvent, they show the above mentioned self-cleaving process yielding the corresponding amine hydrobromide (4a,b) and the cyclic carbonate 4,4-dimethyl-1,3-dioxolan-2-one (5). As explained above, the protecting group undergoes a polar solvent driven intramolecular nucleophilic displacement (Figure 1). In contrast, in nonpolar solvents such as benzene, toluene, chloroform (CHCl3) or dichloromethane (CH2Cl2) 3a,b are very stable, whereas in polar protic solvents such as ethanol (EtOH) or methanol (MeOH) the cleaving takes place very fast (Scheme 2). This deprotecting process can be directly followed using 1H NMR spectroscopy.
![[1860-5397-12-26-i2]](/bjoc/content/inline/1860-5397-12-26-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Stability of 3 in different solvents.
Scheme 2: Stability of 3 in different solvents.
Carpino assumed that, changing one or both of the methyl groups to bulkier substituents would significantly increase the cleaving process [10]. Thus, we attempted to replace one methyl group with a phenyl substituent simply by the reaction of 1a with 1-bromo-2-phenylpropan-2-ol (2b). Although spectroscopic examinations gave evidence for the successful formation of the expected carbamate 3c, only the corresponding amine hydrobromide 4a was isolated as main product. This means that the amino-protecting group is extremely sensitive to decomposition. A third type of monomer was synthesized by reaction of 1a with 1-bromo-2-methylbut-3-en-2-ol (2c) yielding 2-((1-bromo-2-methylbut-3-en-2-yl)oxycarbonylamino)ethyl acrylate (3d, Scheme 3). Although monomer 3d is stable in nonpolar solvents, the cleaving process could not be followed by 1H NMR spectroscopy because of rapid decomposition in a polar solvent.
![[1860-5397-12-26-i3]](/bjoc/content/inline/1860-5397-12-26-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesized derivative monomers 3c,d.
Scheme 3: Synthesized derivative monomers 3c,d.
Next, classical free radical polymerizations of 3a,b were carried out in toluene as solvent at 50 °C using 2,2'-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) as initiator yielding homopolymers 6a,b. This initiator is known to have a low decomposition temperature of 30 °C in toluene. Additionally, copolymers of 3a and 3b, respectively with N,N-dimethylacrylamide (7) were synthesized yielding 7a,b. It was assumed that, since the cleaving process is coupled with cyclization to create the cyclic carbonate 5, the homopolymer could be self-deactivated due to competing hydrogen-bond interactions of the urethane carbonyl group with neighboring groups. Since the comonomer should act as diluting agent a better access of the carbonyl group to the bromo-substituted carbon atom to fulfill the nucleophilic intramolecular displacement could be expected (Scheme 4).
![[1860-5397-12-26-i4]](/bjoc/content/inline/1860-5397-12-26-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: a) Self deactivation of homopolymer 6a,b due to competing hydrogen interactions in comparison to b) copolymer 7a,b.
Scheme 4: a) Self deactivation of homopolymer 6a,b due to competing hydrogen interactions in comparison to b)...
The kinetic studies of 3a, 6a and 7a are shown in Figure 4. Additionally, Figure 5 shows the 1H NMR spectra of homopolymer 6a and deprotected polymer 8a.
![[1860-5397-12-26-4]](/bjoc/content/figures/1860-5397-12-26-4.png?scale=2.38&max-width=1024&background=FFFFFF)
Figure 4: Kinetic studies of the deprotection of 3a, 6a and 7a.
Figure 4: Kinetic studies of the deprotection of 3a, 6a and 7a.
![[1860-5397-12-26-5]](/bjoc/content/figures/1860-5397-12-26-5.png?scale=2.68&max-width=1024&background=FFFFFF)
Figure 5: 300 MHz 1H NMR spectra of a) 6a in CDCl3 and b) 8a in DMSO-d6.
Figure 5: 300 MHz 1H NMR spectra of a) 6a in CDCl3 and b) 8a in DMSO-d6.
The calculated t0.5 and t0.3 values of deprotection for 3a,b, 6a,b and 7a,b are summarized in Table 1.
As assumed above the molecular dispersed monomer shows the highest reactivity followed by the copolymer. The solvolysis of the protecting group in the homopolymer is relatively slow because of neighboring group formed retarding hydrogen bonds (Scheme 4).
Conclusion
It can be concluded from the above described results that the bromo-tert-butyloxycarbonyl group has a certain potential for the preparation of amino group containing functional (meth)acryl polymers. Since the deprotection takes place under mild conditions in polar solvents, some sensitive components, e.g., drugs, proteins, DNA may be present without being affected.
Experimental
Methods
1H NMR measurements were performed using a Bruker Avance 300 operating at 300 MHz at room temperature. Fourier transformation infrared spectroscopy (FTIR) was performed on a Nicolet 6700 FTIR spectrometer equipped with a diamond single bounce ATR accessory. The measurements were performed in the range of 400–3000 cm−1 at room temperature. Differential scanning calorimetry (DSC) was performed using a Mettler Toledo DSC 822 instrument equipped with a sample robot TSO801RO. The Apparatus was controlled over a temperature range between −50 °C and 350 °C at a heating rate of 15 °C min−1. The Tg values are reported as the average of five measurements using the midpoint method. The THF–GPC System compromises a Scharmbeck SFD degasser (Gastor BG12), a FLOW pump (Intelligent PUMP AL-12) and a Scharmbeck SFD (Model S5200) sampler. A Waters 486 Turnable Absorbance Detector and a Scharmbeck SFD RI 2000 detector were used for detection. A set of columns packed with porous styrene–divinylbenzene-copolymer beads was used for separation of the analytes (MZ Analysentechnik GmbH, 1 × guard column 100 Å, 3 × columns with 10,000, 1,000 und 100 Å). The system was calibrated with polystyrene standards with a molecular range from 575 g/mol to 3,114,000 g/mol. THF was used as eluent at a flow rate of 1 mL min−1.
Materials
Commercial reagents and solvents were purchased from Sigma-Aldrich, Merck and Fluka. If not stated otherwise all chemicals were of analytical grade and were used as received without any further purification. 2-Isocyanatoethyl acrylate and (meth)acrylate were purchased from Shōwa Denkō K.K.
Chloroform-d (99.8 atom % D) was obtained from Deutero GmbH (Germany). All solvents were dried by standard methods. Column chromatography was performed using Acros Organics silica gel 60 (230–400 mesh) and thin layer chromatography (TLC) of the products using Merck silica 60 F254 plates.
1-Bromo-2-methylpropan-2-ol (2a)
In a 500 mL three-necked flask with a reflux condenser tert-butanol (100 g, 1.35 mol) was heated under reflux. A second 500 mL single-necked flask was equipped with N-bromosuccinimide (NBS, 50 g 0.28 mol) in a mixture of THF/H2O 1:2. To the boiling tert-butanol 5 portions of 5 mL each concentrated sulfuric acid were added dropwise every 15 min. The forming isobutene was bubbled into the NBS solution under vigorous stirring. After 45 min, all of the NBS disappeared and stirring was continued for an additional hour. THF was evaporated and the remaining aqueous mixture was extracted several times with diethyl ether. The extract was dried over magnesium sulfate and the solution was evaporated. The remaining oil was distilled to give 2a as a pure colorless liquid: yield 22 g (51%); bp 55 °C at 25 mbar; 1H NMR (300 MHz, CDCl3) δ [ppm] 3.39 (s, 2H, -CH2-Br); 2.43 (s, 1H, -OH); 1.30 (s, 6H, -CH3); IR (diamond) ![[Graphic 1]](/bjoc/content/inline/1860-5397-12-26-i5.svg?max-width=637&scale=1.18182) [cm−1]: 3380 (s, νOH), 2976 (m, νC-H), 2931 (m, νC-H), 2872 (m, νC-H), 1465 (m, ν-CH2-), 1379 (s, νOH).
[cm−1]: 3380 (s, νOH), 2976 (m, νC-H), 2931 (m, νC-H), 2872 (m, νC-H), 1465 (m, ν-CH2-), 1379 (s, νOH).
1-Bromo-2-phenylpropan-2-ol (2b)
Into a stirring solution of N-bromosuccinimide (NBS, 32 g, 0.18 mol) in 1/3 THF and 2/3 H2O, α-methylstyrene (10 mL, 0.15 mol) were added. After 2 h of vigorous stirring, THF was evaporated and the aqueous solution was extracted several times with diethyl ether. The extract was dried over magnesium sulfate and the solution was evaporated. The crude product was purified by column chromatography using n-hexane/ethyl acetate 1:1 to give the desired colorless liquid 2b: yield 21.35 g (66%). 1H NMR (300 MHz, CDCl3) δ [ppm] 7.57–7.54 (m, 2H, ArH), 7.38–7.32 (m, 2H, ArH), 7.27–7.22 (m, 1H, ArH), 5.40 (s, 1H, -OH), 3.78 (s, 2H, -CH2-Br), 1.66 (s, 3H, -CH3); IR (diamond) [cm−1]: 3442 (s, νOH), 3027 (m, νC-H), 2978 (m, νC-H), 1602 (w, νC=C), 1493 (w, νC=C), 1446 (m, ν-CH2-), 1374 (s, νOH tert-alcohol).
1-Bromo-2-methylbut-3-en-2-ol (2c)
Into a stirring solution of N-bromosuccinimide (NBS, 20 g, 0.12 mol) in 1/3 THF and 2/3 H2O, isoprene was added in 3 small portions of 5 mL (0.05 mol) at 0 °C. Stirring was continued for an additional hour. THF was evaporated and the remaining aqueous mixture was extracted several times with diethyl ether. The extract was dried over magnesium sulfate and the solution was evaporated. The remaining oil was distilled to give 2c as a pure colorless liquid: yield 7.41 g (37.5%); bp 62 °C at 20 mbar. 1H NMR (300 MHz, CDCl3) δ [ppm] 5.88 (dd, 1H, -CH=CHH), 5.34–5.16 (m, 2H, -CH=CH2), 3.44 (s, 2H, -CH2-Br), 2.32 (s, 1H, -OH), 1.40 (s, 3H, -CH3); IR (diamond) [cm−1]: 3412 (s, νOH), 2980 (m, νC-H), 2928 (m, νC-H), 1644 (m, νC=C), 1453 (m, ν-CH2-), 1371 (s, νOH).
General information for the preparation of the monomers
All syntheses were carried out under an argon atmosphere at room temperature using dibutyltin dilaurate (DBTL) as catalyst.
2-((1-Bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl acrylate (3a)
To a stirring solution of 2-isocyanatoethyl acrylate (1a, 8 g, 0.056 mol) and 2 mol % DBTL in 25 mL toluene, 1-bromo-2-methylpropan-2-ol (2a, 7.7 g, 0.05 mol) was added. After 24 h the solvent was evaporated and the crude product was purified by column chromatography using n-hexane/ethyl acetate 1:1 to give the pure product 3a: yield 5.39 g (32%). 1H NMR (300 MHz, CDCl3) δ [ppm] 6.32 (dd, 3J = 17.3 Hz, 2J = 1.5 Hz, 1H, -CH=CHH), 6.04 (dd, 3J = 17.3 Hz, 3J = 10.4 Hz, 1H, -CH=CH2), 5.76 (dd, 3J = 10.4 Hz, 2J = 1.5 Hz, 1H, -CH=CHH), 5.22 (s, 1H, -NH-), 4.13 (t, 3J = 5.4 Hz, 2H, -OCH2), 3.67 (s, 2H, -CH2-Br), 3.34 (q, 3J = 5.6 Hz, 2H, -NH-CH2), 1.44 (s, 6H, -CH3); IR (diamond) [cm−1]: 3358 (m, νN-H), 2982 (m, νC-H), 2936 (m, νC-H), 1708 (w, νC=O), 1635 (w, νC=C), 1618 (w, νC=Ct), 1516 (s, νN-H), 983 (s, νCH=CH2), 809 (s, νCH=CH2).
2-((1-Bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl methacrylate (3b)
To a stirring solution of 2-isocyanatoethyl methacrylate (1b, 6 g, 0.039 mol) and 2 mol % DBTL in 25 mL toluene, 1-bromo-2-methylpropan-2-ol (2a, 5.8 g, 0.038 mol) was added. After 24 h the solvent was evaporated and the crude product was purified by column chromatography using n-hexane/ethyl acetate 1:1 to give the pure product 3b: yield 5.3 g (43%). 1H NMR (300 MHz, CDCl3) δ [ppm] 6.12–6.11 (m, 1H, -C(CH3)-CHH), 5.59–5.57 (m, 1H, -C(CH3)=CHH), 4.95 (s, 1H, -NH-), 4.21 (t, 3J = 5.4 Hz, 2H, -OCH2), 3.75 (s, 2H,-CH2-Br), 3.44 (m, 2H, -NH-CH2-), 1.93 (s, 3H, -CH3), 1.53 (s, 6H, -CH3); IR (diamond) [cm−1]: 3365 (m, νN-H), 2978 (m, νC-H), 2929 (m, νC-H), 1708 (w, νC=O), 1637 (w, νC=C), 1520 (s, νN-H), 943 (s, νCH=CH2), 814 (s, νCH=CH2).
2-((1-Bromo-2-methylbut-3-en-2-yl)oxycarbonylamino)ethyl acrylate (3d)
To a stirring solution of 2-isocyanatoethyl acrylate (1a, 4.27 g, 0.03 mol) and 2 mol % DBTL in 25 mL toluene, 1-bromo-2-methylbut-3-en-2-ol (2c, 5 g, 0.03 mol) was added. After 24 h the solvent was evaporated and the crude product was purified by column chromatography using n-hexane/ethyl acetate 1:1 to give the pure product 3d: yield 1.7 g (17%). 1H NMR (300 MHz, CDCl3) δ [ppm] 6.43 (dd, 3J = 17,3 Hz, 2J = 1.5 Hz, 1H, -CH-CHH ), 6.12 (dd, 3J = 17.3 Hz, 2J = 10.4 Hz, 2H, -CH-CH2), 5.86 (dd, 3J = 10.4 Hz, 2J = 1.5 Hz, 1H, -CH-CHH), 5.81–5.74 (m, 1H, -CH=CH2), 5.06 (s, 1H, -NH-), 4.58–4.46 (m, 2H, -CH=CH2), 4.24 (t, 2H, -OCH2), 4.01–3.98 (m, 2H, -CH2-Br), 3.50 (q, 2H, -NH-CH2), 1.73 (s, 3H, -CH3); IR (diamond) [cm−1]: 3348 (m, νN-H), 2951 (m, νC-H), 2869 (m, νC-H), 1705 (w, νC=O), 1636 (w, νC=C), 1615 (w, νC=C), 1528 (s, νN-H), 983 (s, νCH=CH2), 808 (s, νCH=CH2).
General information for the preparation of the polymers and copolymers
All polymerizations were carried out in toluene (75 wt %) at 50 °C using 5 mol % 2,2'-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) as initiator. After 24 h the polymerizations were stopped and the solutions were precipitated by pouring into n-hexane. The obtained polymers were filtered off and dried under vacuum.
Poly-(2-((1-bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl acrylate) (6a)
To a mixture of 3a (0.35 g, 1.19 mmol) in 0.7 g toluene, V-70 (5 mol %, 18.35 mg) dissolved in 0.35 g toluene was added. Polymer 6a was obtained as a colorless solid: yield 0.28 g (80%). 1H NMR (300 MHz, CDCl3) δ [ppm] 6.04–5.45 (m, 1H), 4.33–3.97 (m, 2H), 3.90–3.60 (m, 2H), 3.55–3.24 (m, 2H), 2.53–2.12 (m, 1H), 2.09–1.66 (m, 2H), 1.61–1.54 (m, 6H); IR (diamond) [cm−1]: 3359 (m, νN-H), 2955 (m, νC-H), 2930 (m, νC-H), 1698 (w, νC=O), 1516 (s, νN-H); DSC: Tg = 93.1 °C; GPC (THF): ![[Graphic 2]](/bjoc/content/inline/1860-5397-12-26-i6.svg?max-width=637&scale=1.18182) = 9300 g/mol,
= 9300 g/mol, ![[Graphic 3]](/bjoc/content/inline/1860-5397-12-26-i7.svg?max-width=637&scale=1.18182) = 4400 g/mol, D = 2.11.
= 4400 g/mol, D = 2.11.
Poly-(2-((1-bromo-2-methylpropan-2-yl)oxycarbonylamino)ethyl methacrylate) (6b)
To a mixture of 3b (0.38 g, 1.23 mmol) in 0.76 g toluene, V-70 (5 mol %, 19.02 mg) dissolved in 0.38 g toluene was added. Polymer 6b was obtained as a colorless solid: yield 0.34 g (89%). 1H NMR (300 MHz, CDCl3, rt) δ [ppm] 6.12–5.27 (m, 1H), 4.12–3.92 (m, 2H), 3.85–3.65 (m, 2H), 3.55–3.27 (m, 2H), 1.90–1.72 (m, 3H), 1.68–1.60 (m, 2H), 1.58–1.52 (m, 6H); IR (diamond) [cm−1]: 3353 (m, νN-H), 2978 (m, νC-H), 2934 (m, νC-H), 1715 (w, νC=O), 1521 (s, νN-H), 1455 (s, νC-H (R-CH3)); DSC: Tg = 125.9 °C; GPC (THF): = 8900 g/mol, = 6000 g/mol, D = 1.48.
References
-
Burkhart, A.; Fischer, J.; Mondrzyk, A.; Ritter, H. Macromol. Chem. Phys. 2014, 215, 421–425. doi:10.1002/macp.201300734
Return to citation in text: [1] -
Mather, B. D.; Viswanathan, K.; Miller, K. M.; Long, T. E. Prog. Polym. Sci. 2006, 31, 487–531. doi:10.1016/j.progpolymsci.2006.03.001
Return to citation in text: [1] -
Thöne, J.; Ritter, H. Macromol. Chem. Phys. 1987, 188, 2047–2059. doi:10.1002/macp.1987.021880904
Return to citation in text: [1] -
Rehse, H.; Ritter, H. Makromol. Chem. 1989, 190, 697–706. doi:10.1002/macp.1989.021900403
Return to citation in text: [1] -
Gormanns, M.; Rehse, H.; Ritter, H. Makromol. Chem. 1991, 192, 745–755. doi:10.1002/macp.1991.021920401
Return to citation in text: [1] -
Gormanns, M.; Ritter, H. Tetrahedron 1993, 49, 6965–6974. doi:10.1016/S0040-4020(01)87972-2
Return to citation in text: [1] -
Gormanns, M.; Ritter, H. Macromolecules 1994, 27, 5227–5228. doi:10.1021/ma00096a057
Return to citation in text: [1] -
Carpino, L. A.; Parameswaran, K. N.; Kirkley, R. K.; Spiewak, J. W.; Schmitz, E. J. Org. Chem. 1970, 35, 3291–3295. doi:10.1021/jo00835a025
Return to citation in text: [1] -
Carpino, L. A. Acc. Chem. Res. 1973, 6, 191–198. doi:10.1021/ar50066a003
Return to citation in text: [1] -
Carpino, L. A.; Rice, N. W.; Mansour, E. M. E.; Triolo, S. A. J. Org. Chem. 1984, 49, 836–842. doi:10.1021/jo00179a017
Return to citation in text: [1] [2] -
Ohnishi, T.; Sugano, H.; Miyoshi, M. Bull. Chem. Soc. Jpn. 1972, 45, 2603–2607. doi:10.1246/bcsj.45.2603
Return to citation in text: [1]
| 1. | Burkhart, A.; Fischer, J.; Mondrzyk, A.; Ritter, H. Macromol. Chem. Phys. 2014, 215, 421–425. doi:10.1002/macp.201300734 |
| 11. | Ohnishi, T.; Sugano, H.; Miyoshi, M. Bull. Chem. Soc. Jpn. 1972, 45, 2603–2607. doi:10.1246/bcsj.45.2603 |
| 8. | Carpino, L. A.; Parameswaran, K. N.; Kirkley, R. K.; Spiewak, J. W.; Schmitz, E. J. Org. Chem. 1970, 35, 3291–3295. doi:10.1021/jo00835a025 |
| 9. | Carpino, L. A. Acc. Chem. Res. 1973, 6, 191–198. doi:10.1021/ar50066a003 |
| 10. | Carpino, L. A.; Rice, N. W.; Mansour, E. M. E.; Triolo, S. A. J. Org. Chem. 1984, 49, 836–842. doi:10.1021/jo00179a017 |
| 3. | Thöne, J.; Ritter, H. Macromol. Chem. Phys. 1987, 188, 2047–2059. doi:10.1002/macp.1987.021880904 |
| 4. | Rehse, H.; Ritter, H. Makromol. Chem. 1989, 190, 697–706. doi:10.1002/macp.1989.021900403 |
| 5. | Gormanns, M.; Rehse, H.; Ritter, H. Makromol. Chem. 1991, 192, 745–755. doi:10.1002/macp.1991.021920401 |
| 6. | Gormanns, M.; Ritter, H. Tetrahedron 1993, 49, 6965–6974. doi:10.1016/S0040-4020(01)87972-2 |
| 7. | Gormanns, M.; Ritter, H. Macromolecules 1994, 27, 5227–5228. doi:10.1021/ma00096a057 |
| 2. | Mather, B. D.; Viswanathan, K.; Miller, K. M.; Long, T. E. Prog. Polym. Sci. 2006, 31, 487–531. doi:10.1016/j.progpolymsci.2006.03.001 |
| 10. | Carpino, L. A.; Rice, N. W.; Mansour, E. M. E.; Triolo, S. A. J. Org. Chem. 1984, 49, 836–842. doi:10.1021/jo00179a017 |
© 2016 Ritter et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)