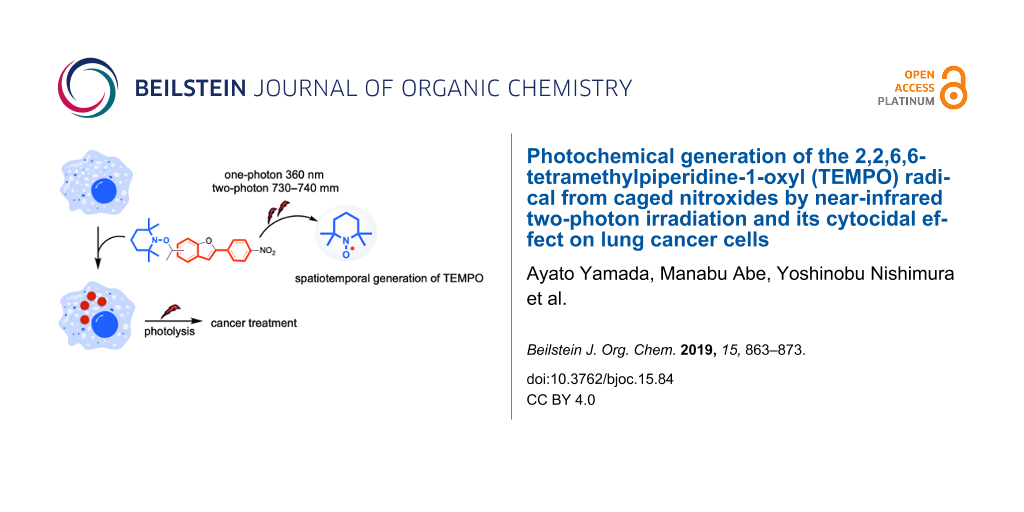
Abstract
Novel caged nitroxides (nitroxide donors) with near-infrared two-photon (TP) responsive character, 2,2,6,6-tetramethyl-1-(1-(2-(4-nitrophenyl)benzofuran-6-yl)ethoxy)piperidine (2a) and its regioisomer 2b, were designed and synthesized. The one-photon (OP) (365 ± 10 nm) and TP (710–760 nm) triggered release (i.e., uncaging) of the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical under air atmosphere were discovered. The quantum yields for the release of the TEMPO radical were 2.5% (2a) and 0.8% (2b) in benzene at ≈1% conversion of 2, and 13.1% (2a) and 12.8% (2b) in DMSO at ≈1% conversion of 2. The TP uncaging efficiencies were determined to be 1.1 GM at 740 nm for 2a and 0.22 GM at 730 nm for 2b in benzene. The cytocidal effect of compound 2a on lung cancer cells under photolysis conditions was also assessed to test the efficacy as anticancer agents. In a medium containing 100 μg mL−1 of 2a exposed to light, the number of living cells decreased significantly compared to the unexposed counterparts (65.8% vs 85.5%).

Graphical Abstract
Introduction
Nitroxides (aminoxyl radicals) possess a delocalized unpaired electron and exhibit negligible dimerization reactivity, making them persistent open-shell species [1-4]. In addition to their ease of handling, nitroxides are highly sensitive to electron paramagnetic resonance (EPR) spectroscopy and redox reactions. Therefore, nitroxides have been developed and utilized in diverse and crucial applications, not only in chemistry, but also in biology, physiology, and energy sciences. These applications include spin-labels [5-7], fluorophore-nitroxide probes [8], contrast agents in magnetic resonance imaging (MRI) [9], polarization transfer agents for nuclear magnetic resonance (NMR) [10-13], and radical batteries [14,15]. Furthermore, the efficient synthesis of polymers with narrow molecular mass distributions has been accomplished using nitroxides as a mediator, i.e., so-called nitroxide-mediated polymerization (NMP) [16-20], and the nitroxide-mediated synthesis of ketones from alcohols is also well utilized in organic synthesis [21-25]. The huge number of studies concerning nitroxides clearly indicates the importance of new methods of generating nitroxides for the future development of science and technology. Notably, in physiological studies [26-32], spatiotemporal control of nitroxide generation is a key approach for investigating the role of redox-active nitroxides in mediating oxidative stress in organisms [27-32].
In 1997, Scaiano and co-workers reported the triplet-xanthone sensitized generation of the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical from alkoxyamine 1 under ultraviolet (355 nm) irradiation (Scheme 1) [33]. De-aerated conditions are necessary for the triplet-sensitized generation of TEMPO due to the triplet quenching ability of O2. The polymerization reactions were initiated via photochemical reaction [34-36]. For physiological studies, however, the photochemical release of nitroxides should be achieved in the presence of O2. Thus, the triplet sensitized method may not be useful for physiological studies. The application of alkoxyamines as theranostic agents [37-40] has been proposed and reported by Brémond and co-workers [41,42].
![[1860-5397-15-84-i1]](/bjoc/content/inline/1860-5397-15-84-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochemical generation of TEMPO radical.
Scheme 1: Photochemical generation of TEMPO radical.
Near-infrared (NIR) photons are excellent light sources in physiological studies as this wavelength of light is less harmful to living tissue than ultraviolet irradiation. Deeper penetration of NIR photons into biological samples is possible using NIR radiation with wavelengths of 650–1050 nm (= 27–44 kcal mol−1). However, in general, chromophores do not absorb at such long wavelengths and the photon energy is too low for bond-cleavage reactions to generate (i.e., uncage) functional molecules. For example, the bond-dissociation energy of the weak PhCH2–OPh, linkage is reported to be 52.1 kcal mol−1 [43]. These issues can be solved by using the NIR-two-photon (TP) excitation technique [44], in which a molecule is electronically excited to the same state generated by one-photon (OP) excitation in the UV–vis region [45]. In addition to the advantages of TP excitation, three-dimensional control of the electronic excitation is possible because the probability of TP excitation is proportional to the square of the light intensity [46]. The light-induced generation of nitroxides using the TP excitation technique, i.e., the concentration jump of nitroxides, is one promising method of exploring the role of these species in life phenomena [47-54] and of promoting site-selective chemical reactions such as polymerization. Very recently, Guillaneuf and co-workers reported the two-photon-induced release of nitroxides in a materials science study [55].
In the last decade, we developed a TP-responsive photo-labile protecting group [56-58] with simple cyclic stilbene structures such as 2-(4-nitrophenyl)benzofuran (NPBF) that absorb in the NIR region of 710–760 nm for the uncaging of bioactive substances such as glutamate and Ca2+ [59-64]. Herein, we report the synthesis of new caged nitroxides (nitroxide donors) 2a and 2b having the TP-responsive NPBF chromophore and the NIR TP-triggered generation of the 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) radical under atmospheric conditions using these species (Scheme 1). Because free radicals are cytotoxic due to their strong DNA-damaging activity [65], they play important roles as anticancer therapeutic agents [66]. Among the free radicals, nitroxides including the TEMPO radical have unique properties, where they can act not only as radical scavengers, but also as anticancer agents [67]. Due to the unique properties described above, nitroxides are not toxic to normal host cells and exhibit toxicity only to tumor cells. Thus, nitroxides are ideal candidates as anticancer therapeutic agents. Based on this knowledge, the cytocidal effect of the radical released from compound 2a on lung cancer cells was tested in vitro, in addition to the fundamental study.
Results and Discussion
The caged-TEMPOs 2a and 2b were synthesized as shown in Scheme 2. The new compounds, 5-ethyl- and 6-ethyl-2-(4-nitrophenyl)benzofuran (5a and 5b), were synthesized from 1-ethynyl-4-nitrobenzene (4) that was prepared from the commercially available 1-iodo-4-nitrobenzene (3) [68]. The TEMPO moiety was introduced at the benzylic position of 5a and 5b using the copper-catalyzed radical reaction in the presence of tert-butyl hydroperoxide (TBHP) to afford 2a and 2b in 38% and 52% yield, respectively [69]. The caged TEMPOs 2a and 2b were thermally stable in benzene below 320 K (47 °C), as confirmed by electron paramagnetic resonance (EPR) spectroscopic analysis. Significant thermal decomposition of 2a and 2b was observed at ≈340 K (67 °C), as indicated by the typical EPR signals (see Supporting Information File 1, Figure S1).
![[1860-5397-15-84-i2]](/bjoc/content/inline/1860-5397-15-84-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of caged nitroxides 2a and 2b.
Scheme 2: Synthesis of caged nitroxides 2a and 2b.
The photophysical data for the new compounds 2a,b and 5a,b are summarized in Table 1. The absorption maxima of compounds 2 and 5 were observed at ≈370 nm with a molar extinction coefficient ε ≈20000 M−1 cm−1 in both benzene and DMSO. The emission profile showed a significant solvent effect. The fluorescence quantum yields in DMSO of 5a and 5b were determined to be 16.1 and 8.6%, respectively, although no emission was observed from these compounds in non-polar benzene, indicating that the excited state has zwitterionic character. The charge transfer transition was supported by time-dependent density functional theory (TD-DFT) calculations for 5a at the CAM-B3LYP/6-31G(d) level of theory (Supporting Information File 1, Figure S2). The fluorescence quantum yields of caged-TEMPO 2a and 2b were found to be 2.9 and 2.2% in DMSO, which are much smaller than those of 5a and 5b, respectively, suggesting the chemical reactivity of the singlet excited states of 2a and 2b.
Table 1: Photophysical data for 2a, 2b, 5a, and 5b in benzene (DMSO).
| Entry | λabs [nm]a | ε [M−1 cm−1] | λem [nm]b | Φf × 102 c | τ [ps]d | |
| 1 | 2a |
371
(375) |
24800
(23100) |
–
(576) |
≈0.0
(2.9) |
–
(220, 1370)e |
| 2 | 2b |
366
(370) |
23000
(23400) |
–
(564) |
≈0.0
(2.2) |
–
(390, 890)f |
| 3 | 5a |
372
(378) |
23800
(20000) |
–
(577) |
≈0.0
(16.1) |
–
(1430) |
| 4 | 5b |
367
(372) |
22300
(19000) |
–
(563) |
≈0.0
(8.6) |
–
(870) |
aAbsorption maximum of 2a, 2b, 5a, 5b. bEmission maximum of 2a (1.18 × 10−6 M), 2b (1.18 × 10−6 M), 5a (1.16 × 10−6 M), 5b (1.12 × 10−6 M). cFluorescence quantum yields. The standard sample 9,10-diphenylanthracene (Φf = 0.91) was used for determining the quantum yields. dFluorescence lifetime monitored at 560 nm. The concentrations were the same as those used for the fluorescence measurements. eEach contribution is 57% and 43%, respectively. fEach contribution is 70% and 30%, respectively.
Time-correlated single photon counting (TCSPC) measurement was performed at 298 K in DMSO to estimate the fluorescence lifetime (τ) of 2 and 5 (Table 1). Single-exponential decay curves were observed for 5a and 5b, respectively (Supporting Information File 1, Figure S3). The lifetimes determined by single-exponential fitting were 1430 (5a) and 870 ps (5b), respectively (Table 1, entries 3 and 4). Double-exponential decay was, however, observed for the TEMPO-substituted NPBF derivatives 2a and 2b, where the lifetimes were 220 (57%) and 1370 ps (43%) for 2a, and 390 (70%) and 890 ps (30%) for 2b (Table 1, entries 1 and 2). For 2a and 2b, intermolecular charge transfer processes induced by the TEMPO moiety may account for the double-exponential decay curves to some extent.
OP photolysis of 2a (5 mM) was first conducted in benzene at ≈298 K using 365 nm light (6.02 × 1015 photons s−1) under atmospheric conditions (Figure 1). Clean release of the TEMPO radical was confirmed by measuring the electron paramagnetic resonance (EPR) signals of the typical nitroxide, AN = 15.5 G (g = 2.00232, Figure 1 and Figure 2c). The first-order rate constant for generation of TEMPO in the bulk photoreaction was found to be k = 1.6 × 10–5 s−1. The amount of photochemically released TEMPO radical was determined by comparing the EPR intensity with the calibration curve of the standard TEMPO sample (Supporting Information File 1, Figure S4). The chemical yield of TEMPO was 80% after 10 min irradiation in benzene under air atmosphere (Figure 2g). Secondary photoreaction of TEMPO gradually decreased the chemical yield of TEMPO. The quantum yield (Φ) for photochemical release of the TEMPO radical was 2.5% at ≈1% conversion in the photolysis of 2a in benzene under atmospheric conditions. Similar photochemical generation of the TEMPO radical was conducted with 2b (5 mM, Supporting Information File 1, Figure S5 and Figure 2d,h). The clean generation of the TEMPO radical was also observed during photolysis under 365 nm irradiation in benzene at ≈298 K under atmospheric conditions, although the reaction was slower than that of 2a, k = 5.5 x 10–6 s–1; Φ = 0.8% at ≈1% conversion of 2b. However, the chemical yield of TEMPO was also high (81% after 20 min irradiation under the same conditions), although slow photochemical decomposition of TEMPO was observed with prolonged irradiation (Figure 2h). In DMSO, the quantum yield for the formation of TEMPO increased significantly to 13.1% (from 2a) and 12.8% (from 2b) at ≈1% conversion of 2 under atmospheric conditions (Figure 1). The notable effect of the solvent on the TEMPO generation may be due to the increase in the lifetime of the excited states. Photochemical decomposition of TEMPO in DMSO was found to be faster than that in benzene, but the chemical yield of TEMPO (56% from 2a and 58% from 2b after 40 s irradiation) was found to be lower than that obtained in benzene (Figure 1).
![[1860-5397-15-84-1]](/bjoc/content/figures/1860-5397-15-84-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Photochemical generation of TEMPO from 2a and 2b. EPR spectra acquired during the photolysis of 2a (5 mM) in benzene using 365 nm LED light under air atmosphere.
Figure 1: Photochemical generation of TEMPO from 2a and 2b. EPR spectra acquired during the photolysis of 2a ...
![[1860-5397-15-84-2]](/bjoc/content/figures/1860-5397-15-84-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Time profile for photochemical generation of TEMPO radical from 2 (5 mM) at ≈298 K in benzene: (a) from 2a under degassed conditions, (b) from 2b under degassed conditions, (c,g) from 2a under air conditions, (d,h) from 2b under air conditions, (e) from 2a under O2, (f) from 2b under O2.
Figure 2: Time profile for photochemical generation of TEMPO radical from 2 (5 mM) at ≈298 K in benzene: (a) ...
To obtain insight into the mechanism of generation of the TEMPO radical, the photolysis of 2 was conducted under degassed conditions using the freeze-pump-thaw (FPT) method (Figure 2a,b). Interestingly, the generation of the TEMPO radical was highly suppressed under the photolysis conditions (Figure 2a,b). Under air conditions, however, the photochemical release of TEMPO was detected in benzene, as shown in Figure 2c,d. Faster formation of TEMPO was observed when O2 atmosphere was used instead of an air atmosphere (Figure 2e,f). Therefore, the O2 molecule may play an important role in clean generation of the TEMPO radical during photolysis. Indeed, the compounds oxidized at the benzylic carbon, 6 and 7, were isolated in 15% (15%) and 56% (42%) yield in the photolysis of 2a and 2b under atmospheric conditions, respectively (Scheme 3), indicating that under degassed conditions, the photochemically generated radical pair returns to the starting compound 2 with rapid radical recombination. Over 70% of the caged TEMPO 2a and ≈85% of 2b were recovered after 2 h of irradiation under degassed conditions. The retarded formation of TEMPO after 5 min of irradiation is due to the decrease in the relative absorbance of 2a to those of primary photoproducts (Figure 2c,e).
![[1860-5397-15-84-i3]](/bjoc/content/inline/1860-5397-15-84-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photochemical generation of TEMPO radical and photoproducts 6 and 7 under air atmosphere.
Scheme 3: Photochemical generation of TEMPO radical and photoproducts 6 and 7 under air atmosphere.
The TP photolysis of 2a (10 mM) and 2b (10 mM) was carried out in benzene under atmospheric conditions using 710, 720, 730, 740, 750, and 760 nm near infrared light from a Ti:sapphire laser (pulse width 100 fs, 80 MHz) emitting at an average of 700 mW (Figure 3 for 2a and Supporting Information File 1, Figure S6 for 2b). The typical EPR signals of TEMPO were also observed after TP excitation of 2a and 2b (Supporting Information File 1, Figure S7). The formation of TEMPO at 740 nm, k740 = 4.9 × 10−6 s−1 in the bulk photoreaction, was the fastest in the TP-uncaging reaction of 2a (Figure 3). For the uncaging reaction of 2b, the rate of consumption under 730 nm irradiation, k730 = 1.6 × 10–6 s−1, was larger than those at other wavelengths (Supporting Information File 1, Figure S6).
![[1860-5397-15-84-3]](/bjoc/content/figures/1860-5397-15-84-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Time profile, ln([2a]/[2a]0) versus irradiation time, of two-photon uncaging reaction of TEMPO in the photolysis of 2a in benzene, at wavelengths of 710–760 nm and power of 700 mW.
Figure 3: Time profile, ln([2a]/[2a]0) versus irradiation time, of two-photon uncaging reaction of TEMPO in t...
The TP action spectra of 2a and 2b in benzene are shown in Figure 4, where the spectra were obtained by extrapolation from the absolute TP cross-section of the parent NPBF (18 GM) at 720 nm [58]. The TP cross-section of 2a was higher than that of 2b by ≈15 GM. This higher GM value may be due to the stronger donor–acceptor character of 2a relative to that of 2b, because the electron-donating alkyl group is located at the para-position of the p-nitrophenyl group in 2a.
![[1860-5397-15-84-4]](/bjoc/content/figures/1860-5397-15-84-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ESR spectra acquired during the photolysis of 2a (5 mM) in benzene using 365 nm light.
Figure 4: ESR spectra acquired during the photolysis of 2a (5 mM) in benzene using 365 nm light.
As observed in the OP uncaging reaction at 365 nm, the efficiency of the TP-induced TEMPO uncaging reaction of 2a was almost three times higher than that of 2b in benzene. This is attributed to the substituent effect of the meta-alkoxy group on the reactivity in the electronically excited states [70]. Moreover, the relative stability of radicals BRa and BRb generated by the photolysis of 2a and 2b had an important impact on the uncaging efficiency. The isodesmic reaction shown in Scheme 4 suggests the radical BRa derived from 2a was 2.04 kcal mol−1 more stable than BRb generated from 2b based on DFT calculations at the B3LYP/6-31G(d) level of theory. In DMSO, no significant difference between 2a and 2b was observed for the photochemical release of the TEMPO species, although the solvent effect is not clearly explained.
![[1860-5397-15-84-i4]](/bjoc/content/inline/1860-5397-15-84-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Isodesmic reaction from BRa and 5b to 5a and BRb.
Scheme 4: Isodesmic reaction from BRa and 5b to 5a and BRb.
As mentioned above, the spatiotemporally controlled generation of the radical pair of TEMPO and BR was confirmed in the photolysis of compounds 2a and 2b. Because free radicals play important roles as anticancer therapeutic agents, the cytocidal effect of the radical released from compound 2a was also tested in vitro using lung cancer cells. One hundred thousand Lewis lung carcinoma (LLC) cells were seeded into 24-well plates (medium: DMEM) and incubated overnight at 37 °C under an atmosphere of 95% air and 5% CO2. The medium was replaced with fresh phenol-red free DMEM containing various concentrations of 2a (0, 10, 100 μg mL−1) and further incubated for 4 h under the same conditions. Without exposure to light, 2a itself exhibited slight cytotoxicity based on trypan blue exclusion, and ≈80–90% living cells remained in the medium containing of 100 μg mL−1 of 2a (Supporting Information File 1, Figure S8).
The cytocidal effect of the radicals released from compound 2a on LLC cells was also tested. Four hours after 1 min exposure to 360 nm light in various concentrations of 2a-containing medium, the number of living cells decreased in a 2a concentration-dependent manner (Supporting Information File 1, Figure S9). After exposure of the cells in the medium containing 100 μg mL−1 of 2a, the number of living cells decreased significantly compared to that without exposure (66.5% vs 87.8%, Supporting Information File 1, Figure S10). An irradiation-time-dependent decline in the viability of the LLC cells was also observed (Figure 5). To evaluate whether the cytocidal effect was due to photochemical radical generation, cells exposed to 360 nm light for 1 min and the unexposed congeners were stained by using a ROS-ID oxidative stress detection kit (Enzo Life Sciences, Farmingdale, NY, U.S.A.). Reactive oxygen species (ROS) were detected in the cells irradiated in the 2a-containing medium, but not in the non-irradiated cells in 2a-containing medium or the irradiated cells without 2a-containing medium (Figure 6). Thus, the preliminary analyses indicated that the photochemical generation of radicals from 2a induced cancer cell death in vitro, although no in vivo study was performed because of the low water solubility of 2a. At this point, we cannot rule out generate of ROS by photosensitization of the chromophore in the presence of O2 for the cytotoxicity.
![[1860-5397-15-84-5]](/bjoc/content/figures/1860-5397-15-84-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Irradiation time-dependent decline in viability of LLC cells with compound 2a.
Figure 5: Irradiation time-dependent decline in viability of LLC cells with compound 2a.
![[1860-5397-15-84-6]](/bjoc/content/figures/1860-5397-15-84-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Detection of intracellular ROS only in irradiated LLC cells with 2a-containing medium.
Figure 6: Detection of intracellular ROS only in irradiated LLC cells with 2a-containing medium.
Conclusion
In the present study, novel caged nitroxides 2a and 2b having a TP-responsive chromophore were synthesized, and OP- and TP-induced generation of the TEMPO radical with these species was examined. The quantum yields for generation of the TEMPO radical from 2a and 2b were determined to be 2.5% and 0.8% in benzene, respectively. The quantum yields in DMSO were found to be higher than those in benzene, 13.1% and 12.8%, respectively. The OP-uncaging efficiency (ε × Φ) was found to be 480 and 175 for 2a and 2b, respectively, at 360 ± 10 nm, in benzene, and 3026 and 2995 in DMSO, respectively. The TP efficiency of the TEMPO uncaging reaction was found to be 1.1 GM at 740 nm for 2a and 0.22 GM at 730 nm for 2b in benzene. The TP-induced clean release of the TEMPO radical is expected to be applicable to further physiological studies and site-selective polymerization reactions.
Experimental
All reagents were purchased from commercial sources and were used without additional purification, unless otherwise mentioned. Caged nitroxides 2a and 2b were prepared according to the methods described previously (Scheme 2) and were isolated by silica gel column chromatography and GPC. 1H and 13C NMR spectra were reported in parts per million (δ) by using CDCl3. IR spectra were recorded with a FTIR spectrometer. UV–vis spectra were taken by a SHIMADZU UV-3600 Plus spectrophotometer. Mass spectra were measured by a Mass Spectrometric Thermo Fisher Scientific LTQ Orbitrap XL, performed by the Natural Science Center for Basic Research and Development (N-BARD), Hiroshima University.
Preparation of caged compounds 2a and 2b
6-Ethyl-2-(4-nitrophenyl)benzofuran (5a). 4-Nitro-1-iodobenzene (16.3 g, 65.5 mmol), Pd(dppf)Cl2 (0.97 g, 1.3 mmol), PPh3 (recrystallized, 0.51 g, 1.9 mmol) and CuI (0.25 g, 1.3 mmol) were added under N2 atmosphere followed by toluene (97 mL) and iPr2NH (49 mL, 359 mmol). The mixture was stirred for 10 min, and TMSA (11.5 mL, 81.6 mmol) in toluene (64 mL) was added at room temperature. It was stirred until all iodobenzene was consumed (20 min). TBAF 1 M in THF (100 mL, 100 mmol) was added followed by 5-ethyl-2-iodophenol (21.6 g, 86.9 mmol). The temperature was increased to 80 °C, and the mixture was stirred for 21.5 h. The reaction was quenched with 10% aqueous citric acid (400 mL) and extracted with DCM. The combined extracts were washed with 10% aqueous NaOH (400 mL), water and dried with anhydrous MgSO4. The solvent was removed by rotary evaporation and the crude product was purified by silica gel column chromatography (hexane/EtOAc 10:1, v/v) to give 6-ethyl-2-(4-nitrophenyl)benzofuran (5a, 10.0 g, 57.3%). mp 114 °C; IR (KBr, cm−1): 3429, 2968, 1601, 1520, 1344, 1108, 828, 825, 754, 692; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.30 (d, J = 9.0 Hz, 2H), 7.98(d, J = 9.0 Hz, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.39 (s, 1H), 7.20 (d, J = 0.9 Hz, 1H), 7.14 (dd, J = 8.0, 1.4 Hz, 1H), 2.80 (q, J = 7.6 Hz, 2H), 1.32 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 156.05 (C), 152.86 (C), 147.12 (C), 143.06 (C), 136.56 (C), 126.43 (C), 125.01 (CH), 124.33 (CH), 124.07 (CH), 121.22 (CH), 110.42 (CH), 105.12 (CH), 29.25 (CH2), 15.84 (CH3); HRMS–ESI (m/z): [M−] calcd. for C16H13NO3, 267.09009; found, 267.09064.
2,2,6,6-Tetramethyl-1-(1-(2-(4-nitrophenyl)benzofuran-6-yl)ethoxy)piperidine (2a). Under air, TEMPO (0.23 g, 1.5 mmol), 6-ethyl-2-(4-nitrophenyl)benzofuran (5a, 1.26 g, 4.72 mmol), Cu(OAc)2 (16.5 mg, 0.092 mmol), bpy (13.8 mg, 0.094 mmol), TBHP (aqueous 70%, 0.41 mL, 2.9 mmol) were added into a two-necked flask in the dark. The reaction was stirred at 60 °C for 15 h. Upon completion, the mixture was purified silica gel column chromatography (hexane/ether 15:1, v/v) to give 2a (236 mg, 37.8%). mp 109 °C; IR (KBr, cm−1): 3429, 2934, 1600, 1521, 1345, 1062, 825; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.31 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.57 (d, J = 8.1 Hz, 1H), 7.54 (s, 1H), 7.25 (dd, J = 8.1, 1.2 Hz, 1H), 7.21 (d, J = 0.7 Hz, 1H), 4.91 (q, J = 6.7 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H), 1.51 (s, 3H), 1.32 (s, 6H), 1.20 (s, 3H), 1.05 (s, 3H), 0.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 155.69 (C), 153.25 (C), 147.18 (C), 144.70 (C), 136.50 (C), 127.39 (C), 125.08 (CH), 124.33 (CH), 122.65 (CH), 121.04 (CH), 109.47 (CH), 105.13 (CH), 83.28 (CH), 59.79 (C), 40.43 (CH2), 34.23 (CH3) , 23.82 (CH2), 20.40 (CH3), 17.24 (CH3); HRMS–ESI (m/z): [M + H]+ calcd. for C25H30N2O4, 423.22783; found, 423.22754.
5-Ethyl-2-(4-nitrophenyl)benzofuran (5b). 4-Nitro-1-iodobenzene (16.8 g, 67.5 mmol), Pd(dppf)Cl2 (1.0 g, 1.3 mmol), PPh3 (recrystallized, 0.53 g, 2.0 mmol) and CuI (0.26 g, 1.3 mmol) were added under N2 atmosphere followed by toluene (100 mL) and iPr2NH (50.5 mL, 370 mmol). The mixture was stirred for 10 min, and TMSA (1.75 mL, 12.5 mmol) in toluene (10 mL) was added at room temperature. It was stirred until all iodobenzene was consumed (20 min). TBAF 1 M in THF (100 mL, 100 mmol) was added followed by 4-ethyl-2-iodophenol (22.22 g, 89.6 mmol). The temperature was increased to 80 °C, and the mixture was stirred for 20 h. The reaction was quenched with 10% aqueous citric acid (666 mL) and extracted with DCM. Combined extracts were washed with 10% aqueous NaOH (666 mL), water and dried with anhydrous MgSO4. The solvent was removed by rotary evaporation and the crude product was purified silica gel column chromatography (hexane/EtOAc 10:1, v/v) to give 5-ethyl-2-(4-nitrophenyl)benzofuran (5b, 12.2 g, 67.6%). mp 129 °C; IR (KBr, cm−1): 2922, 1601, 1514, 1340, 1194, 853, 811, 754, 690; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.30 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.46 (d, J = 9.2 Hz, 1H), 7.44 (s, 1H), 7.21 (dd, J = 8.4, 1.8 Hz, 1H), 7.19 (d, J = 0.8 Hz, 1H), 2.76 (q, J = 7.6 Hz, 2H) 1.30 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 154.12 (C), 153.38 (C), 147.21 (C), 139.74 (C), 136.47 (C), 128.79 (C), 126.24 (CH), 125.12 (CH), 124.29 (CH), 120.14 (CH), 111.12 (CH), 105.04 (CH), 28.83 (CH2), 16.15 (CH3); HRMS–ESI (m/z): [M−] calcd. for C16H13NO3, 267.09009; found, 267.09030.
2,2,6,6-Tetramethyl-1-(1-(2-(4-nitrophenyl)benzofuran-5-yl)ethoxy)piperidine (2b). Under air, TEMPO (46.8 mg, 0.3 mmol), 5-ethyl-2-(4-nitrophenyl)benzofuran (5b, 267 mg, 1 mmol), Cu(OAc)2 (3.6 mg, 0.02 mmol), bpy (3.1 mg, 0.02 mmol), TBHP (aqueous 70%, 0.086 mL, 0.6 mmol) were added into a Schlenk tube in the dark. The reaction was stirred at 60 °C for 16.5 h. Upon completion, the mixture was purified by silica gel column chromatography (hexane/ether 15:1, v/v) to give 2b (66 mg, 52%). mp 144 °C; IR (KBr, cm−1): 2922, 1602, 1520, 1342, 1108, 852, 746; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.31 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.57 (d, J = 1.6 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.36 (dd, J = 8.6, 1.7 Hz, 1H), 7.23 (s, 1H), 4.88 (q, J = 6.6 Hz, 1H), 1.53 (d, J = 6.7 Hz, 3H), 1.51 (s, 3H), 1.33 (s, 6H), 1.19 (s, 3H), 1.02 (s, 3H), 0.60 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 154.75 (C), 153.43 (C), 147.24 (C), 141.55 (C), 136.47 (C), 128.34 (C), 125.15 (CH), 125.04 (CH), 124.32 (CH), 119.50 (CH), 110.98 (CH), 105.39 (CH), 83.13 (CH), 59.76 (C), 40.42 (CH2), 34.37 (CH3), 23.78 (CH2), 20.38 (CH3), 17.24 (CH3); HRMS–ESI (m/z): [M + H]+ calcd. for C25H30N2O4; 423.22783; found 423.22757.
1-(2-(4-Nitrophenyl)benzofuran-6-yl)ethan-1-ol (6a). mp 133 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.31 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.61 (d, J = 7.7 Hz, 1H), 7.61 (s, 1H), 7.30 (dd, J = 8.1, 1.3 Hz, 1H), 7.22 (s, 1H), 5.06 (m, 1H), 1.90 (d, J = 3.6 Hz, 1H), 1.57 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 155.72 (C), 153.61 (C), 147.24 (C), 144.47 (C), 136.26 (C), 127.96 (C), 125.16 (CH), 124.34 (CH), 121.56 (CH), 121.34 (CH), 108.33 (CH), 104.97 (CH), 70.50 (CH), 25.58 (CH3).
1-(2-(4-Nitrophenyl)benzofuran-5-yl)ethan-1-ol (6b). mp 149 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.30 (d, J = 9.0 Hz, 2H), 7.99 (d, J = 9.0 Hz, 2H), 7.66 (s, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.39 (dd, J = 8.5, 1.7 Hz, 1H), 7.22 (s, 1H), 5.04 (q, J = 6.4 Hz, 1H), 1.88 (s, 1H), 1.57 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 154.93 (C), 153.81 (C), 147.29 (C), 141.48 (C), 136.22 (C), 128.74 (C), 125.24 (CH), 124.32 (CH), 123.67 (CH), 118.28 (CH), 111.45 (CH), 105.14 (CH), 70.47 (CH), 25.64 (CH3); HRMS–ESI (m/z): [M−] calcd. for C16H13NO4, 283.08501; found, 283.08548.
1-(2-(4-Nitrophenyl)benzofuran-6-yl)ethan-1-one (7a). mp 214 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.35 (d, J = 8.9 Hz, 2H), 8.18 (s, 1H), 8.05 (d, J = 8.9 Hz, 1H), 7.93 (dd, J = 8.2, 1.3 Hz, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.29 (s, 1H), 2.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.22 (C), 156.45 (C), 155.12 (C), 147.82 (C), 135.52 (C), 134.84 (C), 132.93 (C), 125.76 (CH), 124.41 (CH), 123.88 (CH), 121.41 (CH), 111.79 (CH), 104.85 (CH), 26.86 (CH3).
1-(2-(4-Nitrophenyl)benzofuran-5-yl)ethan-1-one (7b). mp 229 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.34 (d, J = 9.0 Hz, 2H), 8.30 (s, 1H), 8.04 (d, J = 8.6 Hz, 1H), 8.03 (d, J = 9.0 Hz, 2H), 7.62 (d, J = 8.7, 1H), 7.32 (s, 1H), 2.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 197.35 (C), 157.84 (C), 154.86 (C), 147.63 (C), 135.59 (C), 133.41 (C), 128.79 (C), 126.38 (CH), 125.51 (CH), 124.40 (CH), 122.86 (CH), 111.57 (CH), 105.38 (CH), 26.82 (CH3); HRMS–ESI (m/z): [M−] calcd. for C16H11NO4, 281.06936; found, 281.06970.
Photoirradiation of LLC cells with compound 2a
One hundred thousand Lewis lung carcinoma (LLC) cells were seeded into a 24-well plate (medium: DMEM) and incubated overnight at 37 °C in an atmosphere of 95% air and 5% CO2. The medium was replaced with fresh phenol-red free DMEM containing 100 µg/mL of compound 2a. Four hours after various irradiation time of 360 nm light (0, 10, 30, 60, 90, 120, and 150 s) using a fluorescence microscope (BIOREVO BZ-9000, Keyence, Osaka, Japan), cell viability was determined by trypan blue exclusion. Bars represent the mean ± standard deviation (n = 4).
Detection of intracellular ROS in irradiated LLC cells with 2a-containing medium
Fifty thousand Lewis lung carcinoma (LLC) cells were seeded into 24-well plate (medium: DMEM) and incubated overnight at 37 °C in an atmosphere of 95% air and 5% CO2. The medium was replaced with fresh phenol-red free DMEM containing 0 or 100 µg/mL of compound 2a. Thirty minutes after 1 min or no exposure of 360 nm light using a fluorescence microscope (BIOREVO BZ-9000, Keyence, Osaka, Japan), intracellular ROS were detected using the ROS-ID Oxidative Stress Detection Kit (Enzo Life Sciences, Farmingdale, NY, USA) in conjunction with fluorescence microscopy. Intracellular ROS was detected in the form of green fluorescence signals. Bars, 100 µm.
Supporting Information
| Supporting Information File 1: 1H and 13C NMR charts for new compounds and Figures S1–S8. | ||
| Format: PDF | Size: 898.1 KB | Download |
Acknowledgements
The NMR and MS measurements were performed at N-BARD, Hiroshima University. This work was supported by a Grant-in-Aid for Scientific Research JP17H0302200 from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was partly supported by a research grant from Takahashi Industrial and Economic Research Foundation.
References
-
Keana, J. F. W. Chem. Rev. 1978, 78, 37–64. doi:10.1021/cr60311a004
Return to citation in text: [1] -
Janzen, E. G. Acc. Chem. Res. 1971, 4, 31–40. doi:10.1021/ar50037a005
Return to citation in text: [1] -
Eckert, H. Chem. – Eur. J. 2017, 23, 5893–5914. doi:10.1002/chem.201604685
Return to citation in text: [1] -
Krumkacheva, O.; Bagryanskaya, E. J. Magn. Reson. 2017, 280, 117–126. doi:10.1016/j.jmr.2017.02.015
Return to citation in text: [1] -
Kim, N.-K.; Murali, A.; DeRose, V. J. Chem. Biol. 2004, 11, 939–948. doi:10.1016/j.chembiol.2004.04.013
Return to citation in text: [1] -
Sicoli, G.; Wachowius, F.; Bennati, M.; Höbartner, C. Angew. Chem., Int. Ed. 2010, 49, 6443–6447. doi:10.1002/anie.201000713
Return to citation in text: [1] -
Weinrich, T.; Jaumann, E. A.; Scheffer, U. M.; Prisner, T. F.; Göbel, M. W. Beilstein J. Org. Chem. 2018, 14, 1563–1569. doi:10.3762/bjoc.14.133
Return to citation in text: [1] -
Kakavandi, R.; Ravat, P.; Savu, S.-A.; Borozdina, Y. B.; Baumgarten, M.; Casu, M. B. ACS Appl. Mater. Interfaces 2015, 7, 1685–1692. doi:10.1021/am508854u
Return to citation in text: [1] -
Krishna, M. C.; English, S.; Yamada, K.; Yoo, J.; Murugesan, R.; Devasahayam, N.; Cook, J. A.; Golman, K.; Ardenkjaer-Larsen, J. H.; Subramanian, S.; Mitchell, J. B. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 2216–2221. doi:10.1073/pnas.042671399
Return to citation in text: [1] -
Hall, D. A.; Maus, D. C.; Gerfen, G. J.; Inati, S. J.; Becerra, L. R.; Dahlquist, F. W.; Griffin, R. G. Science 1997, 276, 930–932. doi:10.1126/science.276.5314.930
Return to citation in text: [1] -
Matsuki, Y.; Maly, T.; Ouari, O.; Karoui, H.; Le Moigne, F.; Rizzato, E.; Lyubenova, S.; Herzfeld, J.; Prisner, T.; Tordo, P.; Griffin, R. G. Angew. Chem., Int. Ed. 2009, 48, 4996–5000. doi:10.1002/anie.200805940
Return to citation in text: [1] -
Bagryanskaya, E. G.; Marque, S. R. A. Chem. Rev. 2014, 114, 5011–5056. doi:10.1021/cr4000946
Return to citation in text: [1] -
Abe, M. Chem. Rev. 2013, 113, 7011–7088. doi:10.1021/cr400056a
Return to citation in text: [1] -
Nishide, H.; Oyaizu, K. Science 2008, 319, 737–738. doi:10.1126/science.1151831
Return to citation in text: [1] -
Oltean, V.-A.; Renault, S.; Valvo, M.; Brandell, D. Materials 2016, 9, No. 142. doi:10.3390/ma9030142
Return to citation in text: [1] -
Kermagoret, A.; Gigmes, D. Tetrahedron 2016, 72, 7672–7685. doi:10.1016/j.tet.2016.07.002
Return to citation in text: [1] -
Guégain, E.; Guillaneuf, Y.; Nicolas, J. Macromol. Rapid Commun. 2015, 36, 1227–1247. doi:10.1002/marc.201500042
Return to citation in text: [1] -
Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Prog. Polym. Sci. 2013, 38, 63–235. doi:10.1016/j.progpolymsci.2012.06.002
Return to citation in text: [1] -
Bertin, D.; Gigmes, D.; Marque, S. R. A.; Tordo, P. Chem. Soc. Rev. 2011, 40, 2189–2198. doi:10.1039/c0cs00110d
Return to citation in text: [1] -
Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101, 3661–3688. doi:10.1021/cr990119u
Return to citation in text: [1] -
Huple, D. B.; Ghorpade, S.; Liu, R.-S. Adv. Synth. Catal. 2016, 358, 1348–1367. doi:10.1002/adsc.201600018
Return to citation in text: [1] -
Ciriminna, R.; Palmisano, G.; Pagliaro, M. ChemCatChem 2015, 7, 552–558. doi:10.1002/cctc.201402896
Return to citation in text: [1] -
Cao, Q.; Dornan, L. M.; Rogan, L.; Hughes, N. L.; Muldoon, M. J. Chem. Commun. 2014, 50, 4524–4543. doi:10.1039/c3cc47081d
Return to citation in text: [1] -
Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116–3134. doi:10.1039/c3gc41459k
Return to citation in text: [1] -
Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034–5068. doi:10.1002/anie.201002547
Return to citation in text: [1] -
Goeldner, M.; Givens, R., Eds. Dynamic Studies in Biology; Wiley-VCH: Weinheim, 2005.
Return to citation in text: [1] -
Soule, B. P.; Hyodo, F.; Matsumoto, K.; Simone, N. L.; Cook, J. A.; Krishna, M. C.; Mitchell, J. B. Free Radical Biol. Med. 2007, 42, 1632–1650. doi:10.1016/j.freeradbiomed.2007.02.030
Return to citation in text: [1] [2] -
Hoye, A. T.; Davoren, J. E.; Wipf, P.; Fink, M. P.; Kagan, V. E. Acc. Chem. Res. 2008, 41, 87–97. doi:10.1021/ar700135m
Return to citation in text: [1] [2] -
Oliveira, C.; Benfeito, S.; Fernandes, C.; Cagide, F.; Silva, T.; Borges, F. Med. Res. Rev. 2018, 38, 1159–1187. doi:10.1002/med.21461
Return to citation in text: [1] [2] -
Matsuoka, Y.; Yamato, M.; Yamada, K.-i. J. Clin. Biochem. Nutr. 2016, 58, 16–22. doi:10.3164/jcbn.15-105
Return to citation in text: [1] [2] -
Wilcox, C. S. Pharmacol. Ther. 2010, 126, 119–145. doi:10.1016/j.pharmthera.2010.01.003
Return to citation in text: [1] [2] -
Prescott, C.; Bottle, S. E. Cell Biochem. Biophys. 2017, 75, 227–240. doi:10.1007/s12013-016-0759-0
Return to citation in text: [1] [2] -
Scaiano, J. C.; Connolly, T. J.; Mohtat, N.; Pliva, C. N. Can. J. Chem. 1997, 75, 92–97. doi:10.1139/v97-014
Return to citation in text: [1] -
Guillaneuf, Y.; Bertin, D.; Gigmes, D.; Versace, D.-L.; Lalevée, J.; Fouassier, J.-P. Macromolecules 2010, 43, 2204–2212. doi:10.1021/ma902774s
Return to citation in text: [1] -
Versace, D.-L.; Lalevée, J.; Fouassier, J.-P.; Gigmes, D.; Guillaneuf, Y.; Bertin, D. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2910–2915. doi:10.1002/pola.24071
Return to citation in text: [1] -
Yoshida, E. Colloid Polym. Sci. 2010, 288, 1639–1643. doi:10.1007/s00396-010-2287-6
Return to citation in text: [1] -
Kelkar, S. S.; Reineke, T. M. Bioconjugate Chem. 2011, 22, 1879–1903. doi:10.1021/bc200151q
Return to citation in text: [1] -
Melancon, M. P.; Zhou, M.; Li, C. Acc. Chem. Res. 2011, 44, 947–956. doi:10.1021/ar200022e
Return to citation in text: [1] -
Chen, G.; Qiu, H.; Prasad, P. N.; Chen, X. Chem. Rev. 2014, 114, 5161–5214. doi:10.1021/cr400425h
Return to citation in text: [1] -
Lim, E.-K.; Kim, T.; Paik, S.; Haam, S.; Huh, Y.-M.; Lee, K. Chem. Rev. 2015, 115, 327–394. doi:10.1021/cr300213b
Return to citation in text: [1] -
Audran, G.; Brémond, P.; Franconi, J.-M.; Marque, S. R. A.; Massot, P.; Mellet, P.; Parzy, E.; Thiaudière, E. Org. Biomol. Chem. 2014, 12, 719–723. doi:10.1039/c3ob42076k
Return to citation in text: [1] -
Audran, G.; Brémond, P.; Marque, S. R. A. Chem. Commun. 2014, 50, 7921–7928. doi:10.1039/c4cc01364f
Return to citation in text: [1] -
Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, U.S.A., 2002; p 215.
Return to citation in text: [1] -
Göppert-Mayer, M. Ann. Phys. (Berlin, Ger.) 1931, 401, 273–294. doi:10.1002/andp.19314010303
Return to citation in text: [1] -
Boyd, M. Non-Linear Optics, 2nd ed.; Elsevier: London, 2003.
Return to citation in text: [1] -
Kawata, S.; Sun, H.-B.; Tanaka, T.; Takada, K. Nature 2001, 412, 697–698. doi:10.1038/35089130
Return to citation in text: [1] -
Momotake, A.; Lindegger, N.; Niggli, E.; Barsotti, R. J.; Ellis-Davies, G. C. R. Nat. Methods 2006, 3, 35–40. doi:10.1038/nmeth821
Return to citation in text: [1] -
Matsuzaki, M.; Ellis-Davies, G. C. R.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Nat. Neurosci. 2001, 4, 1086–1092. doi:10.1038/nn736
Return to citation in text: [1] -
Mayer, G.; Heckel, A. Angew. Chem., Int. Ed. 2006, 45, 4900–4921. doi:10.1002/anie.200600387
Return to citation in text: [1] -
Ellis-Davies, G. C. R. ACS Chem. Neurosci. 2011, 2, 185–197. doi:10.1021/cn100111a
Return to citation in text: [1] -
Bort, G.; Gallavardin, T.; Ogden, D.; Dalko, P. I. Angew. Chem., Int. Ed. 2013, 52, 4526–4537. doi:10.1002/anie.201204203
Return to citation in text: [1] -
Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchard-Desce, M. Adv. Mater. (Weinheim, Ger.) 2008, 20, 4641–4678. doi:10.1002/adma.200800402
Return to citation in text: [1] -
Papageorgiou, G.; Ogden, D. C.; Barth, A.; Corrie, J. E. T. J. Am. Chem. Soc. 1999, 121, 6503–6504. doi:10.1021/ja990931e
Return to citation in text: [1] -
Matsuzaki, M.; Hayama, T.; Kasai, H.; Ellis-Davies, G. C. R. Nat. Chem. Biol. 2010, 6, 255–257. doi:10.1038/nchembio.321
Return to citation in text: [1] -
Baron, M.; Morris, J. C.; Telitel, S.; Clément, J.-L.; Lalevée, J.; Morlet-Savary, F.; Spangenberg, A.; Malval, J.-P.; Soppera, O.; Gigmes, D.; Guillaneuf, Y. J. Am. Chem. Soc. 2018, 140, 3339–3344. doi:10.1021/jacs.7b12807
Return to citation in text: [1] -
Klán, P.; Šolomek, T.; Bochet, C. G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Chem. Rev. 2013, 113, 119–191. doi:10.1021/cr300177k
Return to citation in text: [1] -
Jakkampudi, S.; Abe, M. In Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2018, Chapter 13667.
Return to citation in text: [1] -
Chitose, Y.; Abe, M. Design and synthesis of two-photon responsive chromophores for application to uncaging reactions. Photochemistry; The Royal Society of Chemistry, 2018; Vol. 46, pp 219–241. doi:10.1039/9781788013598-00219
Return to citation in text: [1] [2] -
Abe, M.; Chitose, Y.; Jakkampudi, S.; Thuy, P. T. T.; Lin, Q.; Van, B. T.; Yamada, A.; Oyama, R.; Sasaki, M.; Katan, C. Synthesis 2017, 49, 3337–3346. doi:10.1055/s-0036-1590813
Return to citation in text: [1] -
Boinapally, S.; Huang, B.; Abe, M.; Katan, C.; Noguchi, J.; Watanabe, S.; Kasai, H.; Xue, B.; Kobayashi, T. J. Org. Chem. 2014, 79, 7822–7830. doi:10.1021/jo501425p
Return to citation in text: [1] -
Komori, N.; Jakkampudi, S.; Motoishi, R.; Abe, M.; Kamada, K.; Furukawa, K.; Katan, C.; Sawada, W.; Takahashi, N.; Kasai, H.; Xue, B.; Kobayashi, T. Chem. Commun. 2016, 52, 331–334. doi:10.1039/c5cc07664a
Return to citation in text: [1] -
Jakkampudi, S.; Abe, M.; Komori, N.; Takagi, R.; Furukawa, K.; Katan, C.; Sawada, W.; Takahashi, N.; Kasai, H. ACS Omega 2016, 1, 193–201. doi:10.1021/acsomega.6b00119
Return to citation in text: [1] -
Chitose, Y.; Abe, M.; Furukawa, K.; Katan, C. Chem. Lett. 2016, 45, 1186–1188. doi:10.1246/cl.160586
Return to citation in text: [1] -
Chitose, Y.; Abe, M.; Furukawa, K.; Lin, J.-Y.; Lin, T.-C.; Katan, C. Org. Lett. 2017, 19, 2622–2625. doi:10.1021/acs.orglett.7b00957
Return to citation in text: [1] -
Fukuhara, K.; Nakashima, T.; Abe, M.; Masuda, T.; Hamada, H.; Iwamoto, H.; Fujitaka, K.; Kohno, N.; Hattori, N. Free Radical Biol. Med. 2017, 106, 1–9. doi:10.1016/j.freeradbiomed.2017.02.014
Return to citation in text: [1] -
Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Nat. Commun. 2015, 6, No. 6907. doi:10.1038/ncomms7907
Return to citation in text: [1] -
Lewandowski, M.; Gwozdzinski, K. Int. J. Mol. Sci. 2017, 18, No. 2490. doi:10.3390/ijms18112490
Return to citation in text: [1] -
Bosiak, M. J. ACS Catal. 2016, 6, 2429–2434. doi:10.1021/acscatal.6b00190
Return to citation in text: [1] -
Li, L.; Yu, Z.; Shen, Z. Adv. Synth. Catal. 2015, 357, 3495–3500. doi:10.1002/adsc.201500544
Return to citation in text: [1] -
Zimmerman, H. E. J. Am. Chem. Soc. 1995, 117, 8988–8991. doi:10.1021/ja00140a014
Return to citation in text: [1]
| 58. | Chitose, Y.; Abe, M. Design and synthesis of two-photon responsive chromophores for application to uncaging reactions. Photochemistry; The Royal Society of Chemistry, 2018; Vol. 46, pp 219–241. doi:10.1039/9781788013598-00219 |
| 70. | Zimmerman, H. E. J. Am. Chem. Soc. 1995, 117, 8988–8991. doi:10.1021/ja00140a014 |
| 1. | Keana, J. F. W. Chem. Rev. 1978, 78, 37–64. doi:10.1021/cr60311a004 |
| 2. | Janzen, E. G. Acc. Chem. Res. 1971, 4, 31–40. doi:10.1021/ar50037a005 |
| 3. | Eckert, H. Chem. – Eur. J. 2017, 23, 5893–5914. doi:10.1002/chem.201604685 |
| 4. | Krumkacheva, O.; Bagryanskaya, E. J. Magn. Reson. 2017, 280, 117–126. doi:10.1016/j.jmr.2017.02.015 |
| 10. | Hall, D. A.; Maus, D. C.; Gerfen, G. J.; Inati, S. J.; Becerra, L. R.; Dahlquist, F. W.; Griffin, R. G. Science 1997, 276, 930–932. doi:10.1126/science.276.5314.930 |
| 11. | Matsuki, Y.; Maly, T.; Ouari, O.; Karoui, H.; Le Moigne, F.; Rizzato, E.; Lyubenova, S.; Herzfeld, J.; Prisner, T.; Tordo, P.; Griffin, R. G. Angew. Chem., Int. Ed. 2009, 48, 4996–5000. doi:10.1002/anie.200805940 |
| 12. | Bagryanskaya, E. G.; Marque, S. R. A. Chem. Rev. 2014, 114, 5011–5056. doi:10.1021/cr4000946 |
| 13. | Abe, M. Chem. Rev. 2013, 113, 7011–7088. doi:10.1021/cr400056a |
| 43. | Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, U.S.A., 2002; p 215. |
| 9. | Krishna, M. C.; English, S.; Yamada, K.; Yoo, J.; Murugesan, R.; Devasahayam, N.; Cook, J. A.; Golman, K.; Ardenkjaer-Larsen, J. H.; Subramanian, S.; Mitchell, J. B. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 2216–2221. doi:10.1073/pnas.042671399 |
| 44. | Göppert-Mayer, M. Ann. Phys. (Berlin, Ger.) 1931, 401, 273–294. doi:10.1002/andp.19314010303 |
| 8. | Kakavandi, R.; Ravat, P.; Savu, S.-A.; Borozdina, Y. B.; Baumgarten, M.; Casu, M. B. ACS Appl. Mater. Interfaces 2015, 7, 1685–1692. doi:10.1021/am508854u |
| 37. | Kelkar, S. S.; Reineke, T. M. Bioconjugate Chem. 2011, 22, 1879–1903. doi:10.1021/bc200151q |
| 38. | Melancon, M. P.; Zhou, M.; Li, C. Acc. Chem. Res. 2011, 44, 947–956. doi:10.1021/ar200022e |
| 39. | Chen, G.; Qiu, H.; Prasad, P. N.; Chen, X. Chem. Rev. 2014, 114, 5161–5214. doi:10.1021/cr400425h |
| 40. | Lim, E.-K.; Kim, T.; Paik, S.; Haam, S.; Huh, Y.-M.; Lee, K. Chem. Rev. 2015, 115, 327–394. doi:10.1021/cr300213b |
| 5. | Kim, N.-K.; Murali, A.; DeRose, V. J. Chem. Biol. 2004, 11, 939–948. doi:10.1016/j.chembiol.2004.04.013 |
| 6. | Sicoli, G.; Wachowius, F.; Bennati, M.; Höbartner, C. Angew. Chem., Int. Ed. 2010, 49, 6443–6447. doi:10.1002/anie.201000713 |
| 7. | Weinrich, T.; Jaumann, E. A.; Scheffer, U. M.; Prisner, T. F.; Göbel, M. W. Beilstein J. Org. Chem. 2018, 14, 1563–1569. doi:10.3762/bjoc.14.133 |
| 41. | Audran, G.; Brémond, P.; Franconi, J.-M.; Marque, S. R. A.; Massot, P.; Mellet, P.; Parzy, E.; Thiaudière, E. Org. Biomol. Chem. 2014, 12, 719–723. doi:10.1039/c3ob42076k |
| 42. | Audran, G.; Brémond, P.; Marque, S. R. A. Chem. Commun. 2014, 50, 7921–7928. doi:10.1039/c4cc01364f |
| 26. | Goeldner, M.; Givens, R., Eds. Dynamic Studies in Biology; Wiley-VCH: Weinheim, 2005. |
| 27. | Soule, B. P.; Hyodo, F.; Matsumoto, K.; Simone, N. L.; Cook, J. A.; Krishna, M. C.; Mitchell, J. B. Free Radical Biol. Med. 2007, 42, 1632–1650. doi:10.1016/j.freeradbiomed.2007.02.030 |
| 28. | Hoye, A. T.; Davoren, J. E.; Wipf, P.; Fink, M. P.; Kagan, V. E. Acc. Chem. Res. 2008, 41, 87–97. doi:10.1021/ar700135m |
| 29. | Oliveira, C.; Benfeito, S.; Fernandes, C.; Cagide, F.; Silva, T.; Borges, F. Med. Res. Rev. 2018, 38, 1159–1187. doi:10.1002/med.21461 |
| 30. | Matsuoka, Y.; Yamato, M.; Yamada, K.-i. J. Clin. Biochem. Nutr. 2016, 58, 16–22. doi:10.3164/jcbn.15-105 |
| 31. | Wilcox, C. S. Pharmacol. Ther. 2010, 126, 119–145. doi:10.1016/j.pharmthera.2010.01.003 |
| 32. | Prescott, C.; Bottle, S. E. Cell Biochem. Biophys. 2017, 75, 227–240. doi:10.1007/s12013-016-0759-0 |
| 33. | Scaiano, J. C.; Connolly, T. J.; Mohtat, N.; Pliva, C. N. Can. J. Chem. 1997, 75, 92–97. doi:10.1139/v97-014 |
| 21. | Huple, D. B.; Ghorpade, S.; Liu, R.-S. Adv. Synth. Catal. 2016, 358, 1348–1367. doi:10.1002/adsc.201600018 |
| 22. | Ciriminna, R.; Palmisano, G.; Pagliaro, M. ChemCatChem 2015, 7, 552–558. doi:10.1002/cctc.201402896 |
| 23. | Cao, Q.; Dornan, L. M.; Rogan, L.; Hughes, N. L.; Muldoon, M. J. Chem. Commun. 2014, 50, 4524–4543. doi:10.1039/c3cc47081d |
| 24. | Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116–3134. doi:10.1039/c3gc41459k |
| 25. | Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034–5068. doi:10.1002/anie.201002547 |
| 34. | Guillaneuf, Y.; Bertin, D.; Gigmes, D.; Versace, D.-L.; Lalevée, J.; Fouassier, J.-P. Macromolecules 2010, 43, 2204–2212. doi:10.1021/ma902774s |
| 35. | Versace, D.-L.; Lalevée, J.; Fouassier, J.-P.; Gigmes, D.; Guillaneuf, Y.; Bertin, D. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 2910–2915. doi:10.1002/pola.24071 |
| 36. | Yoshida, E. Colloid Polym. Sci. 2010, 288, 1639–1643. doi:10.1007/s00396-010-2287-6 |
| 16. | Kermagoret, A.; Gigmes, D. Tetrahedron 2016, 72, 7672–7685. doi:10.1016/j.tet.2016.07.002 |
| 17. | Guégain, E.; Guillaneuf, Y.; Nicolas, J. Macromol. Rapid Commun. 2015, 36, 1227–1247. doi:10.1002/marc.201500042 |
| 18. | Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Prog. Polym. Sci. 2013, 38, 63–235. doi:10.1016/j.progpolymsci.2012.06.002 |
| 19. | Bertin, D.; Gigmes, D.; Marque, S. R. A.; Tordo, P. Chem. Soc. Rev. 2011, 40, 2189–2198. doi:10.1039/c0cs00110d |
| 20. | Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101, 3661–3688. doi:10.1021/cr990119u |
| 14. | Nishide, H.; Oyaizu, K. Science 2008, 319, 737–738. doi:10.1126/science.1151831 |
| 15. | Oltean, V.-A.; Renault, S.; Valvo, M.; Brandell, D. Materials 2016, 9, No. 142. doi:10.3390/ma9030142 |
| 27. | Soule, B. P.; Hyodo, F.; Matsumoto, K.; Simone, N. L.; Cook, J. A.; Krishna, M. C.; Mitchell, J. B. Free Radical Biol. Med. 2007, 42, 1632–1650. doi:10.1016/j.freeradbiomed.2007.02.030 |
| 28. | Hoye, A. T.; Davoren, J. E.; Wipf, P.; Fink, M. P.; Kagan, V. E. Acc. Chem. Res. 2008, 41, 87–97. doi:10.1021/ar700135m |
| 29. | Oliveira, C.; Benfeito, S.; Fernandes, C.; Cagide, F.; Silva, T.; Borges, F. Med. Res. Rev. 2018, 38, 1159–1187. doi:10.1002/med.21461 |
| 30. | Matsuoka, Y.; Yamato, M.; Yamada, K.-i. J. Clin. Biochem. Nutr. 2016, 58, 16–22. doi:10.3164/jcbn.15-105 |
| 31. | Wilcox, C. S. Pharmacol. Ther. 2010, 126, 119–145. doi:10.1016/j.pharmthera.2010.01.003 |
| 32. | Prescott, C.; Bottle, S. E. Cell Biochem. Biophys. 2017, 75, 227–240. doi:10.1007/s12013-016-0759-0 |
| 47. | Momotake, A.; Lindegger, N.; Niggli, E.; Barsotti, R. J.; Ellis-Davies, G. C. R. Nat. Methods 2006, 3, 35–40. doi:10.1038/nmeth821 |
| 48. | Matsuzaki, M.; Ellis-Davies, G. C. R.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Nat. Neurosci. 2001, 4, 1086–1092. doi:10.1038/nn736 |
| 49. | Mayer, G.; Heckel, A. Angew. Chem., Int. Ed. 2006, 45, 4900–4921. doi:10.1002/anie.200600387 |
| 50. | Ellis-Davies, G. C. R. ACS Chem. Neurosci. 2011, 2, 185–197. doi:10.1021/cn100111a |
| 51. | Bort, G.; Gallavardin, T.; Ogden, D.; Dalko, P. I. Angew. Chem., Int. Ed. 2013, 52, 4526–4537. doi:10.1002/anie.201204203 |
| 52. | Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchard-Desce, M. Adv. Mater. (Weinheim, Ger.) 2008, 20, 4641–4678. doi:10.1002/adma.200800402 |
| 53. | Papageorgiou, G.; Ogden, D. C.; Barth, A.; Corrie, J. E. T. J. Am. Chem. Soc. 1999, 121, 6503–6504. doi:10.1021/ja990931e |
| 54. | Matsuzaki, M.; Hayama, T.; Kasai, H.; Ellis-Davies, G. C. R. Nat. Chem. Biol. 2010, 6, 255–257. doi:10.1038/nchembio.321 |
| 46. | Kawata, S.; Sun, H.-B.; Tanaka, T.; Takada, K. Nature 2001, 412, 697–698. doi:10.1038/35089130 |
| 69. | Li, L.; Yu, Z.; Shen, Z. Adv. Synth. Catal. 2015, 357, 3495–3500. doi:10.1002/adsc.201500544 |
| 66. | Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Nat. Commun. 2015, 6, No. 6907. doi:10.1038/ncomms7907 |
| 67. | Lewandowski, M.; Gwozdzinski, K. Int. J. Mol. Sci. 2017, 18, No. 2490. doi:10.3390/ijms18112490 |
| 59. | Abe, M.; Chitose, Y.; Jakkampudi, S.; Thuy, P. T. T.; Lin, Q.; Van, B. T.; Yamada, A.; Oyama, R.; Sasaki, M.; Katan, C. Synthesis 2017, 49, 3337–3346. doi:10.1055/s-0036-1590813 |
| 60. | Boinapally, S.; Huang, B.; Abe, M.; Katan, C.; Noguchi, J.; Watanabe, S.; Kasai, H.; Xue, B.; Kobayashi, T. J. Org. Chem. 2014, 79, 7822–7830. doi:10.1021/jo501425p |
| 61. | Komori, N.; Jakkampudi, S.; Motoishi, R.; Abe, M.; Kamada, K.; Furukawa, K.; Katan, C.; Sawada, W.; Takahashi, N.; Kasai, H.; Xue, B.; Kobayashi, T. Chem. Commun. 2016, 52, 331–334. doi:10.1039/c5cc07664a |
| 62. | Jakkampudi, S.; Abe, M.; Komori, N.; Takagi, R.; Furukawa, K.; Katan, C.; Sawada, W.; Takahashi, N.; Kasai, H. ACS Omega 2016, 1, 193–201. doi:10.1021/acsomega.6b00119 |
| 63. | Chitose, Y.; Abe, M.; Furukawa, K.; Katan, C. Chem. Lett. 2016, 45, 1186–1188. doi:10.1246/cl.160586 |
| 64. | Chitose, Y.; Abe, M.; Furukawa, K.; Lin, J.-Y.; Lin, T.-C.; Katan, C. Org. Lett. 2017, 19, 2622–2625. doi:10.1021/acs.orglett.7b00957 |
| 65. | Fukuhara, K.; Nakashima, T.; Abe, M.; Masuda, T.; Hamada, H.; Iwamoto, H.; Fujitaka, K.; Kohno, N.; Hattori, N. Free Radical Biol. Med. 2017, 106, 1–9. doi:10.1016/j.freeradbiomed.2017.02.014 |
| 55. | Baron, M.; Morris, J. C.; Telitel, S.; Clément, J.-L.; Lalevée, J.; Morlet-Savary, F.; Spangenberg, A.; Malval, J.-P.; Soppera, O.; Gigmes, D.; Guillaneuf, Y. J. Am. Chem. Soc. 2018, 140, 3339–3344. doi:10.1021/jacs.7b12807 |
| 56. | Klán, P.; Šolomek, T.; Bochet, C. G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Chem. Rev. 2013, 113, 119–191. doi:10.1021/cr300177k |
| 57. | Jakkampudi, S.; Abe, M. In Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2018, Chapter 13667. |
| 58. | Chitose, Y.; Abe, M. Design and synthesis of two-photon responsive chromophores for application to uncaging reactions. Photochemistry; The Royal Society of Chemistry, 2018; Vol. 46, pp 219–241. doi:10.1039/9781788013598-00219 |
© 2019 Yamada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)