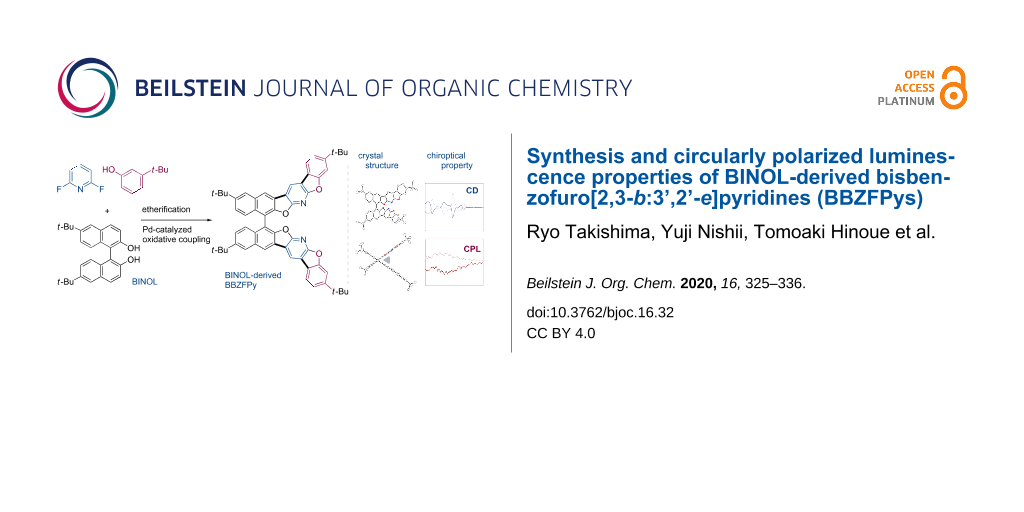
Abstract
A series of optically active bisbenzofuro[2,3-b:3’,2’-e]pyridine (BBZFPy) derivatives was synthesized starting with the readily available (S)- and (R)-1,1’-bi-2-naphthols through a palladium-catalyzed multiple intramolecular C–H/C–H coupling as the key ring-closure step. The effect of terminal tert-butyl substituents on the BBZFPy skeleton was systematically investigated to uncover a unique aggregation-induced enhancement of CPL characteristics in the solid state. The crystal structures of the coupling products were also evaluated by single crystal X-ray analysis and the well-ordered intermolecular stacking arrangements appeared to be responsible for the enhanced CPL.

Graphical Abstract
Introduction
Densely-fused (hetero)aromatic compounds have been a key motif in a wide range of manufactured functional molecules, as they exhibit fundamentally useful electrochemical and photophysical properties. Considerable effort has therefore taken into the development of efficient methods for the construction of such polycyclic scaffolds, and the last decade has witnessed a remarkable improvement in the palladium-catalyzed C–H/C–H oxidative coupling as one of the potential synthetic strategies [1]. This method is straightforward and highly step-economical, enabling us to produce condensed (hetero)acenes from rather simple polyarenes, in which several aromatic units are connected with each other through appropriate linker units [2-11]. Recently, we reported the synthesis and optical properties of a series of furan-fused aromatics via the formal dehydrogenative coupling adopting oxygen atom as the linker [12-17]. In particular, bisbenzofuro[2,3-b:3′,2′-e]pyridines (BBZFPys) were found to exhibit intense photoluminescence with relatively high quantum efficiency (Φflu up to 0.70), indicating that the BBFZPy scaffold may serve as a key fluorophore unit in certain light-emitting functional materials (Scheme 1) [14].
![[1860-5397-16-32-i1]](/bjoc/content/inline/1860-5397-16-32-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of BBFZPys through the Pd-catalyzed C–H/C–H coupling.
Scheme 1: Synthesis of BBFZPys through the Pd-catalyzed C–H/C–H coupling.
Meanwhile, organic optoelectronic materials with circularly polarized luminescence (CPL) characteristics have attracted significant research interests in recent years [18-21] for their potential applications in three-dimensional displays [22], information storage systems [23], molecular photoswitches [24], etc. Among a series of chiral scaffolds for CPL emitting molecules, axially chiral 1,1’-bi-2-naphthol (BINOL) has been frequently adopted for the core structure owing to the availability of both enantiomers as well as the ease of site-selective functionalization. Up to date, many BINOL-based CPL active compounds have been established by installing aromatic subunits on the periphery of the binaphthyl skeleton or on the hydroxy groups [25-32], extending the π-system [33], and linearly connecting the naphthyl rings [34-36]. In these compounds, the hydroxy groups are remained untouched or protected as the corresponding ethers or esters. We envisioned that the assembly of the binaphthyl-fused furan motif embedding the BINOL hydroxy groups into the polyaromatic scaffolds would lead to the development of new chiroptical materials. Such molecules, however, have hardly been investigated to date probably because of the synthetic difficulty to obtain them as pure enantiomers. There have been only a few reports for the binaphthyl-fused furan-ring construction from the C3-alkynylated BINOL derivatives [37-39]. In this context, we herein describe the synthesis of axially chiral BINOL-derived BBZFPys through the palladium-catalyzed oxidative coupling reaction. The optical properties of the synthesized polyaromatic compounds were systematically studied, and some of them displayed an interesting aggregation-induced enhancement of CPL in the solid state with considerably higher dissymmetry factors compared to those in solution.
Results and Discussion
Synthesis of BINOL-derived BBFPys
The study was initiated with the synthesis of 2,6-diaryloxypyridines 3 bearing a 1,1’-binaphthyl backbone as precursors for the dehydrogenative coupling reaction (Scheme 2). In general, functionalization of the BINOL hydroxy groups should be performed at temperatures below 80 °C to prevent racemization [40,41]. 6,6’-Di-tert-butyl-1,1’-bi-2-naphthol (1) was treated with 2,6-difluoropyridine using cesium carbonate as base in DMF at 40 °C [42], giving both the enantiomers of 2 in optically pure forms. The remaining fluorine substituents were subsequently replaced by a series of phenols including unsubstituted phenol, p-tert-butylphenol, and m-tert-butylphenol to produce the corresponding unsymmetrically substituted pyridines 3a–c in high yields. We then examined the oxidative cyclization of these compounds under the standard conditions adopting Pd(TFA)2 (30 mol %, TFA = trifluoroacetate) and AgOAc (3.0 equiv) as catalyst and oxidant, respectively, in pivalic acid as solvent (Scheme 3). Since the desired four-fold coupling products 4 were obtained only in small quantities after the reactions, the crude mixtures containing incompletely cyclized compounds were again subjected to the same catalytic conditions. To our delight, all the target molecules 4a–c were successfully isolated as pure enantiomers in 10–36% yields. The higher yield of 4c was probably due to its better solubility.
![[1860-5397-16-32-i2]](/bjoc/content/inline/1860-5397-16-32-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-32-i3]](/bjoc/content/inline/1860-5397-16-32-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4a–c through oxidative coupling reaction.
Scheme 3: Synthesis of 4a–c through oxidative coupling reaction.
In order to systematically evaluate the optical properties of these coupling products, a simple benzofuran-fused 1,1’-binaphthyl 6 was also synthesized as a benchmark (Scheme 4). The parent ether 5 was obtained through the arylation of 1 utilizing Ph2IOTf as arylating reagent [43,44]. Some copper-mediated arylation protocols using bromobenzene or iodobenzene [45,46] were also applicable to the preparation of 5, but significant loss of optical purity was inevitable. After the Pd-catalyzed cyclization under the standard conditions, the desired compounds (S)- and (R)-6 were obtained as pure enantiomers in 18% yield.
![[1860-5397-16-32-i4]](/bjoc/content/inline/1860-5397-16-32-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Optical properties
We next investigated the optical properties of the coupling products (Figure 1 and Table 1). The parent compounds 3a–c emitted fluorescence at around 360 nm both in sufficiently diluted CHCl3 solutions (1.0 × 10−5 M) and in the solid states. The quantum yields of these molecules were around 0.15 in solution, which is typical for binaphthyl compounds [47]. In contrast, 4a–c as well as 6 exhibited fluorescence at around 390 nm in solution, with relatively higher quantum yields of 0.37–0.40. The emission bands of 4a–c in their solid state were considerably red-shifted as compared to that of 6, suggesting that these compounds displayed the appreciable effect of molecular aggregation. Interestingly, 4b and 4c, bearing the additional terminal tert-butyl substituents, were more red-shifted than 4a despite such a sterically demanding group usually disturbs intermolecular stacking interactions.
![[1860-5397-16-32-1]](/bjoc/content/figures/1860-5397-16-32-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Absorption (dotted line) and fluorescence (solid line) spectra of 3, 4, and 6 measured as CHCl3 solutions (1.0 × 10−5 M) and in solid states.
Figure 1: Absorption (dotted line) and fluorescence (solid line) spectra of 3, 4, and 6 measured as CHCl3 sol...
Table 1: Florescence properties.a
| Compd. | solution λmax (λex) | solid λmax (λex) | Φ (solution) | Φ (solid) |
| 3a | 358 nm (282 nm) | 360 nm (341 nm) | 0.13 | 0.30 |
| 3b | 357 nm (283 nm) | 360 nm (338 nm) | 0.14 | 0.21 |
| 3c | 357 nm (283 nm) | 360 nm (338 nm) | 0.16 | 0.19 |
| 4a | 392 nm (341 nm) | 436 nm (369 nm) | 0.39 | 0.17 |
| 4b | 391 nm (342 nm) | 488 nm (369 nm) | 0.38 | 0.08 |
| 4c | 391 nm (342 nm) | 457 nm (370 nm) | 0.40 | 0.07 |
| 6 | 384 nm (263 nm) | 405 nm (380 nm) | 0.37 | 0.13 |
aMeasured at room temperature as solution in CHCl3 (1.0 × 10−5 M) and in the solid states.
Subsequently, the chiroptical properties of the synthesized compounds were evaluated (Figure 2 and Figure 3, Table 2). The circular dichroism (CD) spectra in CHCl3 solutions showed apparent Cotton signals characteristic to axially chiral molecules. In all cases, the (S)- and (R)-enantiomers were evidently mirror images of each other while the anisotropy factors gabs are relatively small and in the range of 10−4 to 10−7. The spectral shapes of 3a–c and 4a–c were respectively comparable, indicating that the posted positions of the terminal tert-butyl groups exerted minimal influence on the Cotton effect in the solution state. A similar trend was observed for the CPL spectra. The (S)-isomers displayed left-handed CPL characteristics throughout the wavelengths of their corresponding fluorescence emission bands, whereas the (R)-isomers emitted right-handed CPL to produce the mirror images. The calculated luminescence dissymmetry factors [48] glum for the solutions were all within the range of 3.80 × 10−4 to 6.90 × 10−4. On the other hand, in the dispersed solid state in Fomblin® PFPE (perfluoropolyether) fluid (each sample was not soluble in the fluid and gave the expected solid-state luminescence), the signal intensity drastically changed depending on the molecular structures. In particular, 4b and 4c exhibited enhanced CPL characteristics with considerably high glum values of 6.68 × 10−3 and 6.06 × 10−3, respectively, which were approximately ten times larger than those of the CHCl3 solutions [49]. The parent compounds 3a–c, however, did not give clear mirror images in the CPL measurements. Since such a phenomenon was not observed for 4a and 6, the terminal tert-butyl substituents in 4b and 4c were likely to assist the formation of well-ordered aggregates, being consistent with the observation of the red-shifted luminescence discussed above.
![[1860-5397-16-32-2]](/bjoc/content/figures/1860-5397-16-32-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: CD and CPL spectra of 3 measured as CHCl3 solutions (1.0 × 10−5 M) and in the solid states (dispersed in Fomblin®).
Figure 2: CD and CPL spectra of 3 measured as CHCl3 solutions (1.0 × 10−5 M) and in the solid states (dispers...
![[1860-5397-16-32-3]](/bjoc/content/figures/1860-5397-16-32-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CD and CPL spectra of 4 and 6 measured as CHCl3 solutions (1.0 × 10−5 M) and in solid states (dispersed in Fomblin®).
Figure 3: CD and CPL spectra of 4 and 6 measured as CHCl3 solutions (1.0 × 10−5 M) and in solid states (dispe...
Table 2: Calculated dimensionless dissymmetry factors.a
| Compd. | gabs (solution) | glum (solution) | glum (solid) |
| 3a | 8.72 × 10−7 (285 nm) | 4.37 × 10−4 (358 nm) | n.d.b |
| 3b | 3.18 × 10−7 (285 nm) | 6.72 × 10−4 (357 nm) | n.d.b |
| 3c | 1.06 × 10−6 (285 nm) | 6.90 × 10−4 (357 nm) | n.d.b |
| 4a | 5.60 × 10−6 (341 nm) | 5.57 × 10−4 (392 nm) | 5.40 × 10−4 (436 nm) |
| 4b | 9.96 × 10−5 (342 nm) | 4.60 × 10−4 (391 nm) | 6.68 × 10−3 (488 nm) |
| 4c | 8.72 × 10−5 (342 nm) | 6.40 × 10−4 (391 nm) | 6.06 × 10−3 (457 nm) |
| 6 | 1.00 × 10−4 (256 nm) | 3.80 × 10−4 (384 nm) | 1.40 × 10−4 (405 nm) |
aMeasured at room temperature as solution in CHCl3 (1.0 × 10−5 M) and in solid states (dispersed in Fomblin®). bNot determined.
Crystal structures of 4b and 4c
The molecular structures of 4b and 4c were unambiguously determined by single crystal X-ray diffraction analysis. The crystal of 4b is classified into a space group P4322 (tetragonal) with a biaryl torsion angle of 74.4° (Figure 4b). A considerable intermolecular π–π stacking interaction was observed in between its polyaromatic fragments whose distance is approximately 3.44 Å. The polycyclic subunits overlap each other, being line-symmetrically aligned (Figure 5a). Meanwhile, the isomer 4c has two independent molecules in the unit cell, and the torsion angles are 67.1° and 106.8°, respectively (Figure 4d and 4f). As displayed in Figure 4b, the aromatic fragments are point-symmetrically overlapped with the π–π stacking distance of around 3.45 Å. It is noteworthy that both 4b and 4c pile up while minimizing the steric repulsion between the tert-butyl groups which occupy “staggered” orientations in their crystal structures (Figure 5c and 5d). Unfortunately, the crystal structure of 4a was not determined after numerous attempts for obtaining crystals suitable for the X-ray analysis. Based on these observations, it is reasonable to conclude that the tert-butyl substituents effectively restricted the stacking structure to the specific conformations, thereby facilitating the assembly of well-ordered aggregates in the solid state [50-54].
![[1860-5397-16-32-4]](/bjoc/content/figures/1860-5397-16-32-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawings of 4b and 4c with 50% thermal probability. Hydrogen atoms and solvent molecules are omitted for clarity. Only major orientation of the disordered structure is displayed. The CCDC numbers are 1971471 for (R)-4b and 1971470 for (R)-4c.
Figure 4: ORTEP drawings of 4b and 4c with 50% thermal probability. Hydrogen atoms and solvent molecules are ...
![[1860-5397-16-32-5]](/bjoc/content/figures/1860-5397-16-32-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Intramolecular stacking structures of 4b and 4c.
Figure 5: Intramolecular stacking structures of 4b and 4c.
Conclusion
In summary, we have achieved the synthesis of a series of CPL-active polyheteroaromatic compounds from readily available chiral BINOLs via the O-arylation and subsequent palladium-catalyzed C–H/C–H coupling reaction. The substitution pattern on the BBZFPy skeleton had much effect on the solid-state optical properties. Particularly, the compounds 4b and 4c bearing terminal tert-butyl groups exhibited solid-state fluorescence with the enhanced CPL characteristics (glum = 6.68 × 10−3 and 6.06 × 10−3), as compared to those in solution. Their solid-state structures were investigated by X-ray diffraction analysis to find well-ordered intermolecular stacking structures within the crystals.
Experimental
General
All manipulations were performed under N2 using standard Schlenk techniques unless otherwise noted. DMF was dried and deoxygenated by a Glass Counter Solvent Dispending System (Nikko Hansen & Co., Ltd.). DMSO was distilled from CaH2 and stored over molecular sieves 4 Å. Silica gel column chromatography was performed using Wakosil® C-200 (64–210 μm). Nuclear magnetic resonance spectra were measured at 400 MHz (1H NMR) and at 100 MHz (13C NMR) in 5 mm NMR tubes. 1H NMR chemical shifts were reported in ppm relative to the resonance of TMS (δ 0.00) or the residual solvent signals at δ 7.26 for CDCl3. 13C NMR chemical shifts were reported in ppm relative to the residual solvent signals at δ 77.2 for CDCl3. Melting points were measured using a Mettler Toledo MP90. High-resolution mass spectra (HRMS) were recorded by APCI-TOF or EI. Preparative gel permeation chromatography (GPC) was conducted with a YMC GPC-T2000 column eluting with CHCl3. Absorption spectra were recorded with JASCO V-750 spectrometer. Photoluminescence spectra were recorded with JASCO FP-8500 spectrometer. Quantum yield was determined using an integration sphere system. CD and CPL spectra were recorded with JASCO J-820AC and JASCO CPL-300 spectrometers. HPLC analysis was carried out with JASCO EXTREMA (PU4180/MD4015/CO4065) equipped with YMC CHIRAL ART Amylose-SA and YMC CHIRAL ART Cellulose-SB columns.
Preparation of 6,6'-di-tert-butyl-BINOL (1)
Compound 1 was prepared according to a literature procedure [55]. 1H NMR (400 MHz, CDCl3) δ 1.38 (s, 18H), 4.96 (s, 2H), 7.13 (d, J = 8.9 Hz, 2H), 7.36 (d, J = 8.9 Hz, 2H), 7.41 (dd, J = 2.0, 8.9 Hz, 2H), 7.82 (d, J = 2.0 Hz, 2H), 7.94 (d, J = 8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.25, 34.61, 110.62, 117.54, 123.49, 124.01, 126.36, 129.35, 131.34, 131.42, 146.69, 152.29; HRMS–APCI (m/z): [M + H]+ calcd for C28H31O2, 399.2330; found, 399.2319. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/2-propanol 90:10, 1.0 mL/min, 40 °C; (S)-1: tR = 17.9 min, (R)-1: tR = 6.83 min, UV detection at 250.0 nm.
Preparation of 2
To a 20 mL two-necked round-bottomed flask were added 1 (796 mg, 2.0 mmol) and Cs2CO3 (978 mg, 6.0 mmol). 2,6-Difluoropyridine (0.55 mL, 6.0 mmol) and DMF (10 mL) were added via syringe. The mixture was stirred at 40 °C for 48 h under N2. The resulting mixture was extracted with EtOAc. The organic layer was washed with water, dried over Na2SO4, and evaporated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/EtOAc 2:1) and GPC to give the title compound as white solid; (S)-2 (1.01 g, 86% yield), (R)-2 (1.06 g, 90% yield). Mp 212–214 °C; 1H NMR (400 MHz, CDCl3) δ 1.37 (s, 18H), 6.28 (dd, J = 2.6, 7.8 Hz, 2H), 6.37 (dd, J = 1.2, 8.0 Hz, 2H), 7.20 (d, J = 0.8 Hz, 2H), 7.33 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 7.39 (dd, J = 2.0, 8.4 Hz, 2H), 7.77 (d, J = 2.0 Hz, 2H), 7.89 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.18, 34.68, 101.52, 101.87, 121.38, 122.81, 123.20, 125.29, 126.12, 129.51, 131.09, 131.96, 142.60 (d, JC–F = 7.8 Hz), 147.89, 148.95, 161.61 (d, JC–F = 14.0 Hz), 162.28 (d, JC–F = 240 Hz), 19F NMR (376 MHz, CDCl3) δ 69.06; HRMS–APCI (m/z): [M + H]+ calcd for C38H35F2N2O2, 589.2644; found, 589.2661. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Cellulose-SB column, n-hexane/chloroform 95:5, 1.0 mL/min, 40 °C; (S)-2: tR = 9.86 min, (R)-2: tR = 21.29 min, UV detection at 250.0 nm.
Preparation of 3a–c
Compound 3a: To a 10 mL Schlenk flask were added 2 (294 mg, 0.5 mmol), phenol (103 mg, 1.1 mmol), and Cs2CO3 (358 mg, 1.1 mmol). DMSO (3.5 mL) was added via the syringe. The mixture was stirred at 100 °C for 18 h under N2. The resulting mixture was extracted with EtOAc. The organic layer was washed with water, dried over Na2SO4, and evaporated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/EtOAc 6:1) and GPC to give the title compound as white solid; (S)-3a (309 mg, 84% yield), (R)-3a (332 mg, 90%). Mp 110–112 °C; 1H NMR (400 MHz, CDCl3) δ 1.29 (s, 18H), 6.10 (d, J = 7.9 Hz, 2H), 6.16 (d, J = 7.9 Hz, 2H), 6.91 (dd, J = 0.92, 8.5 Hz, 4H), 6.99–7.03 (m, 4H), 7.14–7.17 (m, 8H), 7.22 (d, J = 8.8 Hz, 2H), 7.68 (d, J = 2.0 Hz, 2H), 7.73 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.23, 34.63, 103.72, 104.83, 121.03, 121.55, 122.67, 123.15, 124.27, 125.03, 126.13, 129.10, 129.30, 130.90, 132.04, 141.35, 147.49, 149.54, 154.00, 161.86, 162.48; HRMS–APCI (m/z): [M + H]+ calcd for C50H45N2O4, 737.3367; found, 737.3374. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 95:5, 1.0 mL/min, 40 °C; (S)-3a: tR = 7.25 min, (R)-3a: tR = 14.12 min, UV detection at 250.0 nm.
Compound 3b: Synthesized similarly to 3a using 4-tert-butylphenol. Purified by silica gel column chromatography (eluent: hexane/EtOAc 4:1) and GPC to give the title compound as white solid; (S)-3b (399 mg, 94% yield), (R)-3b (386 mg, 91% yield). Mp 113–115 °C; 1H NMR (400 MHz, CDCl3) δ 1.16 (s, 18H), 1.35 (s, 18H), 6.20 (d, J = 8.0 Hz, 2H), 6.28 (d, J = 7.6 Hz, 2H), 6.77 (d, J = 8.9 Hz, 2H), 6.88 (dd, J = 2.1, 6.7 Hz, 4H), 7.09 (dd, J = 2.0, 8.0 Hz, 2H), 7.12 (dd, J = 2.1, 7.0 Hz, 4H), 7.23 (d, J = 8.8 Hz, 2H), 7.29 (t, J = 8.0 Hz, 2H), 7.72 (d, J = 2.0 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.23, 31.95, 34.22, 34.58, 103.43, 104.29, 120.83, 121.94, 122.40, 123.40, 125.01, 125.73, 126.23, 128.83, 130.79, 132.05, 141.29, 146.89, 147.26, 149.60, 151.15, 161.82, 162.41; HRMS–APCI (m/z): [M + H]+ calcd for C58H61N2O4, 849.4603; found, 849.4626. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 95:5, 1.0 mL/min, 40 °C; (S)-3b: tR = 8.36 min, (R)-3b: tR = 10.05 min, UV detection at 250.0 nm.
Compound 3c: Synthesized similarly to 3a using 3-tert-butylphenol. Purified by silica gel column chromatography (eluent: hexane/EtOAc 6:1) and GPC to give the title compound as white solid; (S)-3c (377 mg, 89% yield), (R)-3c (403 mg, 95% yield). Mp 88–90 °C; 1H NMR (400 MHz, CDCl3) δ 1.17 (s, 18H), 1.36 (s, 18H), 6.14 (d, J = 8.0 Hz, 2H), 6.23 (d, J = 7.6 Hz, 2H), 6.79 (ddd, J = 1.0, 2.2, 7.3 Hz, 2H), 7.02 (t, J = 2.0 Hz, 2H), 7.09–7.13 (m, 4H), 7.16–7.28 (m, 8H), 7.73 (d, J = 1.6 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.16, 31.22, 34.61, 34.63, 103.53, 104.62, 117.99, 118.29, 121.39, 121.53, 122.67, 123.11, 124.94, 126.13, 128.82, 129.06, 130.86, 132.03, 141.26, 147.42, 149.48, 153.01, 153.87, 162.23, 162.50; HRMS–APCI (m/z): [M + H]+ calcd for C58H61N2O4, 849.4599; found, 849.4626. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 95:5, 1.0 mL/min, 40 °C; (S)-3c: tR = 9.00 min, (R)-3c: tR = 8.61 min, UV detection at 250.0 nm.
Preparation of 4a–c
Compound 4a: To a 10 mL Schlenk flask were added 3a (184 mg, 0.25 mmol), Pd(TFA)2 (24.9 mg, 0.075 mmol), AgOAc (167 mg, 1.0 mmol), and PivOH (2.0 mL). The mixture was heated at 150 °C for 24 h under air. After cooling to room temperature, the resulting mixture was diluted with water and filtered through a pad of Celite eluting with dichloromethane. The filtrate was washed with water, dried over Na2SO4, and concentrated in vacuo. The obtained crude material was again subjected to the catalytic conditions described above. The residue was purified by silica gel column chromatography (eluent: hexane/dichloromethane 4:1) and GPC to give the title compound as pale yellow solid; (S)-4a (28.2 mg, 16% yield), (R)-4a (18.1 mg, 10% yield). Mp >300 °C; 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 18H), 7.44–7.52 (m, 8H), 7.62 (d, J = 8.0 Hz, 2H), 8.07 (dd, J = 0.8, 7.6 Hz, 2H), 8.12 (d, J = 2.0 Hz, 2H), 8.67 (s, 2H), 8.96 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 31.25, 34.82, 112.20, 112.62, 113.07, 113.65, 120.13, 120.59, 122.80, 122.93, 123.21, 123.61, 123.62, 125.68, 125.81, 127.39, 130.67, 130.90, 147.553, 151.62, 154.63, 161.90, 163.10; HRMS–APCI (m/z): [M + H]+ calcd for C50H37N2O4, 729.2723; found, 729.2748. The enantiomeric purity was determined by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 60:40, 1.0 mL/min, 40 °C; (S)-4a: tR = 4.39 min, (R)-4a: tR = 6.73 min, UV detection at 250.0 nm.
Compound 4b: Synthesized similarly to 4b from 3b (254 mg, 0.30 mmol). Purified by silica gel column chromatography (eluent: hexane/EtOAc 4:1) and GPC to give the title compound as pale yellow solid; (S)-4b (45.4 mg, 18% yield), (R)-4b (30.2 mg, 12% yield). Single crystals suitable for the X-ray analysis were obtained by slow evaporation from EtOAc solution. Mp >300 °C; 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 18H), 1.48 (s, 18H), 7.47–7.71 (m, 8H), 8.07 (d, J = 0.8 Hz, 2H), 8.11 (d, J = 1.6 Hz, 2H), 8.65 (s, 2H), 8.98 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 31.26, 31.87, 34.82, 34.98, 111.49, 112.62, 113.40, 113.47, 116.98, 119.97, 122.31, 122.75, 122.35, 123.57, 125.13, 125.69, 125.73, 130.64, 130.87, 146.82, 147.48, 151.62, 152.78, 162.25, 162.94; HRMS–EI (m/z): [M]+ calcd for C58H52N2O4, 840.3927; found, 840.3932; ![[Graphic 1]](/bjoc/content/inline/1860-5397-16-32-i5.svg?max-width=637&scale=1.18182) = +31.8 (S-isomer), −32.4 (R-isomer) as CHCl3 solution. The enantiomeric purity was determined by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/2-propanol 90:10, 1.0 mL/min, 40 °C; (S)-4b: tR = 15.95 min, (R)-4b: tR = 24.38 min, UV detection at 250.0 nm.
= +31.8 (S-isomer), −32.4 (R-isomer) as CHCl3 solution. The enantiomeric purity was determined by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/2-propanol 90:10, 1.0 mL/min, 40 °C; (S)-4b: tR = 15.95 min, (R)-4b: tR = 24.38 min, UV detection at 250.0 nm.
Compound 4c: Synthesized similarly to 4c from 3c (254 mg, 0.3 mmol). Purified by silica gel column chromatography (eluent: hexane/EtOAc 4:1) and GPC to give the title compound as pale yellow solid; (S)-4c (74.1 mg, 29% yield), (R)-4c (90.0 mg, 36% yield). Single crystals suitable for the X-ray analysis were obtained by hexane vapor diffusion into CHCl3 solution. Mp >300 °C; 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 1.45 (s, 18H), 7.47–7.64 (m, 6H), 7.637 (d, J = 0.8 Hz, 2H), 7.97 (d, J = 8.4 Hz, 2H), 8.10 (d, J = 0.8 Hz ,2H), 8.65 (s, 2H), 8.91 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 31.26, 31.57, 34.81, 3.38, 109.04, 112.62, 113.23, 113.39, 119.98, 119.99, 120.05, 121.20, 122.53, 123.35, 123.58, 125.69, 125.70, 130.63, 130.88, 147.46, 151.62, 151.76, 155.05, 162.12, 162.77; HRMS–APCI (m/z): [M + H]+ calcd for C58H53N2O4, 841.3970; found, 841.4000. The enantiomeric purity was determined by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 70:30, 1.0 mL/min, 40 °C; (S)-4c: tR = 4.48 min, (R)-4c: tR = 6.66 min, UV detection at 250.0 nm.
Preparation of 5
In a 200 mL three-necked round-bottomed flask, 1 (1.99 g, 5.0 mmol) was added to a suspension of t-BuOK (1.40 g, 12.5 mmol) in THF (80 mL) at 0 °C. After stirring for 2 h, Ph2IOTf (5.38 g, 12.5 mmol) was added in one portion. The mixture was allowed to warm to 40 °C, and stirred at this temperature until the complete consumption 1 was confirmed by TLC. The resulting suspension was poured into ice water and extracted with Et2O. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/EtOAc 20:1) and GPC (CHCl3) to give the title compound as white solid; (S)-5 (1.59 g, 58% yield), (R)-5 (1.71 g, 62% yield). Mp 167.0–169.0 °C; 1H NMR (400 MHz, CDCl3) δ 1.39 (s, 18H), 6.79 (d, J = 7.6, Hz, 4H), 6.92 (t, J = 7.6 Hz, 2H), 7.10 (t, J = 7.6 Hz, 4H), 7.17 (d, J = 8.9 Hz, 2H), 7.27 (d, J = 9.2 Hz, 2H), 7.39 (dd, J = 2.0, 8.9 Hz, 2H), 7.79 (d, J = 1.7 Hz, 2H), 7.83 (d, J = 8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 31.25, 34.65, 118.84, 119.29, 122.06, 122.43, 123.04, 125.46, 125.63, 129.19 129.46, 130.30, 132.44, 147.18, 152.08, 157.74; HRMS–APCI [m/z]: [M + H]+ calcd for C40H39O2, 551.2959; found, 551.2945. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/chloroform 98:2, 1.0 mL/min, 40 °C; (S)-5: tR = 4.91 min, (R)-5: tR = 5.32 min, UV detection at 250.0 nm.
Preparation of 6
To a 10 mL Schlenk flask were added 5 (165 mg, 0.3 mmol), Pd(TFA)2 (29.9 mg, 0.09 mmol), AgOAc (198 mg, 1.2 mmol), and PivOH (3.0 mL). The mixture was heated at 150 °C for 24 h under air. After cooling to room temperature, the resulting mixture was diluted with water and filtered through a pad of Celite eluting with dichloromethane. The filtrate was washed with water, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane/EtOAc 2:1) and GPC to give the title compound as pale yellow solid; (S)-6 (30.1 mg, 18% yield), (R)-6 (30.3 mg, 18% yield). Mp 212–214 °C; 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 18H), 7.31 (dd, J = 0.8, 7.6 Hz, 2H), 7.35 (dd, J = 1.2, 7.6 Hz, 2H), 7.38–7.43 (m, 6H), 8.09 (s, 2H), 8.13–8.16 (m, 2H), 8.61 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 31.26, 34.74, 111.88, 112.25, 119.71, 121.16, 122.62, 123.61, 124.35, 125.08, 125.26, 125.55, 128.08, 130.54, 130.75, 146.80, 153.36, 157.70; HRMS–APCI (m/z): [M + H]+ calcd for C40H35O2, 547.2622; found, 547.2632. The enantiomeric purity was confirmed by HPLC analysis: CHIRAL ART Amylose-SA column, n-hexane/EtOAc 95:5, 1.0 mL/min, 40 °C; (S)-6: tR = 6.47 min, (R)-6: tR = 6.24 min, UV detection at 250.0 nm.
Supporting Information
| Supporting Information File 1: Summary of X-ray crystallography data, copy of NMR spectra, and copy of HPLC charts. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567
See for a recent review on the oxidative coupling.
Return to citation in text: [1] -
Yoshimoto, H.; Itatani, H. Bull. Chem. Soc. Jpn. 1973, 46, 2490–2492. doi:10.1246/bcsj.46.2490
Return to citation in text: [1] -
Shiotani, A.; Itatani, H. Angew. Chem., Int. Ed. Engl. 1974, 13, 471–472. doi:10.1002/anie.197404711
Return to citation in text: [1] -
Åkermark, B.; Eberson, L.; Jonsson, E.; Pettersson, E. J. Org. Chem. 1975, 40, 1365–1367. doi:10.1021/jo00897a048
Return to citation in text: [1] -
Knölker, H.-J.; O'Sullivan, N. Tetrahedron 1994, 50, 10893–10908. doi:10.1016/s0040-4020(01)85701-x
Return to citation in text: [1] -
Åkermark, B.; Oslob, J. D.; Heuschert, U. Tetrahedron Lett. 1995, 36, 1325–1326. doi:10.1016/0040-4039(94)02467-p
Return to citation in text: [1] -
Hagelin, H.; Oslob, J. D.; Åkermark, B. Chem. – Eur. J. 1999, 5, 2413–2416. doi:10.1002/(sici)1521-3765(19990802)5:8<2413::aid-chem2413>3.0.co;2-3
Return to citation in text: [1] -
Knölker, H.-J.; Fröhner, W.; Reddy, K. R. Synthesis 2002, 557–564. doi:10.1055/s-2002-20953
Return to citation in text: [1] -
Matsubara, S.; Asano, K.; Kajita, Y.; Yamamoto, M. Synthesis 2007, 2055–2059. doi:10.1055/s-2007-983738
Return to citation in text: [1] -
Watanabe, T.; Ueda, S.; Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Chem. Commun. 2007, 4516–4518. doi:10.1039/b707899d
Return to citation in text: [1] -
Liégault, B.; Lee, D.; Huestis, M. P.; Stuart, D. R.; Fagnou, K. J. Org. Chem. 2008, 73, 5022–5028. doi:10.1021/jo800596m
Return to citation in text: [1] -
Kaida, H.; Satoh, T.; Hirano, K.; Miura, M. Chem. Lett. 2015, 44, 1125–1127. doi:10.1246/cl.150408
Return to citation in text: [1] -
Kaida, H.; Satoh, T.; Nishii, Y.; Hirano, K.; Miura, M. Chem. Lett. 2016, 45, 1069–1071. doi:10.1246/cl.160496
Return to citation in text: [1] -
Kaida, H.; Goya, T.; Nishii, Y.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2017, 19, 1236–1239. doi:10.1021/acs.orglett.7b00323
Return to citation in text: [1] [2] -
Itai, Y.; Nishii, Y.; Stachelek, P.; Data, P.; Takeda, Y.; Minakata, S.; Miura, M. J. Org. Chem. 2018, 83, 10289–10302. doi:10.1021/acs.joc.8b01451
Return to citation in text: [1] -
Nakamura, S.; Tohnai, N.; Nishii, Y.; Hinoue, T.; Miura, M. ChemPhotoChem 2019, 3, 46–53. doi:10.1002/cptc.201800189
Return to citation in text: [1] -
Nakamura, S.; Okamoto, M.; Tohnai, N.; Nakayama, K.-i.; Nishii, Y.; Miura, M. Bull. Chem. Soc. Jpn. 2020, 93, 99–108. doi:10.1246/bcsj.20190269
Return to citation in text: [1] -
Sánchez-Carnerero, E. M.; Agarrabeitia, A. R.; Moreno, F.; Maroto, B. L.; Muller, G.; Ortiz, M. J.; de la Moya, S. Chem. – Eur. J. 2015, 21, 13488–13500. doi:10.1002/chem.201501178
Return to citation in text: [1] -
Tanaka, H.; Inoue, Y.; Mori, T. ChemPhotoChem 2018, 2, 386–402. doi:10.1002/cptc.201800015
Return to citation in text: [1] -
Chen, N.; Yan, B. Molecules 2018, 23, 3376. doi:10.3390/molecules23123376
Return to citation in text: [1] -
Ma, J.-L.; Peng, Q.; Zhao, C.-H. Chem. – Eur. J. 2019, 25, 15441–15454. doi:10.1002/chem.201903252
Return to citation in text: [1] -
Schadt, M. Annu. Rev. Mater. Sci. 1997, 27, 305–379. doi:10.1146/annurev.matsci.27.1.305
Return to citation in text: [1] -
Sherson, J. F.; Krauter, H.; Olsson, R. K.; Julsgaard, B.; Hammerer, K.; Cirac, I.; Polzik, E. S. Nature 2006, 443, 557–560. doi:10.1038/nature05136
Return to citation in text: [1] -
Feringa, B. L. Acc. Chem. Res. 2001, 34, 504–513. doi:10.1021/ar0001721
Return to citation in text: [1] -
Kawai, T.; Kawamura, K.; Tsumatori, H.; Ishikawa, M.; Naito, M.; Fujiki, M.; Nakashima, T. ChemPhysChem 2007, 8, 1465–1468. doi:10.1002/cphc.200600747
Return to citation in text: [1] -
Amako, T.; Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. Tetrahedron 2013, 69, 2753–2757. doi:10.1016/j.tet.2013.01.084
Return to citation in text: [1] -
Nakabayashi, K.; Amako, T.; Tajima, N.; Fujiki, M.; Imai, Y. Chem. Commun. 2014, 50, 13228–13230. doi:10.1039/c4cc02946a
Return to citation in text: [1] -
Feuillastre, S.; Pauton, M.; Gao, L.; Desmarchelier, A.; Riives, A. J.; Prim, D.; Tondelier, D.; Geffroy, B.; Muller, G.; Clavier, G.; Pieters, G. J. Am. Chem. Soc. 2016, 138, 3990–3993. doi:10.1021/jacs.6b00850
Return to citation in text: [1] -
Kitatobe, T.; Mimura, Y.; Tsujimoto, S.; Tajima, N.; Fujiki, M.; Imai, Y. Tetrahedron 2017, 73, 6856–6862. doi:10.1016/j.tet.2017.10.036
Return to citation in text: [1] -
Zhang, X.; Zhang, Y.; Li, Y.; Quan, Y.; Cheng, Y.; Li, Y. Chem. Commun. 2019, 55, 9845–9848. doi:10.1039/c9cc04289j
Return to citation in text: [1] -
Zhang, X.; Zhang, Y.; Zhang, H.; Quan, Y.; Li, Y.; Cheng, Y.; Ye, S. Org. Lett. 2019, 21, 439–443. doi:10.1021/acs.orglett.8b03620
Return to citation in text: [1] -
Wu, Z.-G.; Han, H.-B.; Yan, Z.-P.; Luo, X.-F.; Wang, Y.; Zheng, Y.-X.; Zuo, J.-L.; Pan, Y. Adv. Mater. (Weinheim, Ger.) 2019, 31, 1900524. doi:10.1002/adma.201900524
Return to citation in text: [1] -
Hassan, K.; Yamashita, K.-i.; Hirabayashi, K.; Shimizu, T.; Nakabayashi, K.; Imai, Y.; Matsumoto, T.; Yamano, A.; Sugiura, K.-i. Chem. Lett. 2015, 44, 1607–1609. doi:10.1246/cl.150704
Return to citation in text: [1] -
Takaishi, K.; Yamamoto, T.; Hinoide, S.; Ema, T. Chem. – Eur. J. 2017, 23, 9249–9252. doi:10.1002/chem.201702143
Return to citation in text: [1] -
Takaishi, K.; Takehana, R.; Ema, T. Chem. Commun. 2018, 54, 1449–1452. doi:10.1039/c7cc09187g
Return to citation in text: [1] -
Takaishi, K.; Iwachido, K.; Takehana, R.; Uchiyama, M.; Ema, T. J. Am. Chem. Soc. 2019, 141, 6185–6190. doi:10.1021/jacs.9b02582
Return to citation in text: [1] -
Droz, A. S.; Neidlein, U.; Anderson, S.; Seiler, P.; Diederich, F. Helv. Chim. Acta 2001, 84, 2243–2289. doi:10.1002/1522-2675(20010815)84:8<2243::aid-hlca2243>3.0.co;2-g
Return to citation in text: [1] -
Ye, P.; Li, Q.; Bai, Z.; Dong, K.; Liu, Q. Heterocycles 2015, 91, 1986–1995. doi:10.3987/com-15-13302
Return to citation in text: [1] -
Octa-Smolin, F.; van der Vight, F.; Yadav, R.; Bhangu, J.; Soloviova, K.; Wölper, C.; Daniliuc, C. G.; Strassert, C. A.; Somnitz, H.; Jansen, G.; Niemeyer, J. J. Org. Chem. 2018, 83, 14568–14587. doi:10.1021/acs.joc.8b02353
Return to citation in text: [1] -
Meca, L.; Řeha, D.; Havlas, Z. J. Org. Chem. 2003, 68, 5677–5680. doi:10.1021/jo034344u
Return to citation in text: [1] -
Moustafa, G. A. I.; Oki, Y.; Akai, S. Angew. Chem., Int. Ed. 2018, 57, 10278–10282. doi:10.1002/anie.201804161
Return to citation in text: [1] -
Paul, A.; Kumar, A.; Nanjunda, R.; Farahat, A. A.; Boykin, D. W.; Wilson, W. D. Org. Biomol. Chem. 2017, 15, 827–835. doi:10.1039/c6ob02390h
Return to citation in text: [1] -
Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t
Return to citation in text: [1] -
Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645
Return to citation in text: [1] -
Sperotto, E.; de Vries, J. G.; van Klink, G. P. M.; van Koten, G. Tetrahedron Lett. 2007, 48, 7366–7370. doi:10.1016/j.tetlet.2007.08.026
Return to citation in text: [1] -
Liu, X.; Zhang, S. Synlett 2011, 268–272. doi:10.1055/s-0030-1259291
Return to citation in text: [1] -
Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. Chem. – Asian J. 2012, 7, 2836–2841. doi:10.1002/asia.201200725
Return to citation in text: [1] -
Emeis, C. A.; Oosterhoff, L. J. J. Chem. Phys. 1971, 54, 4809–4819. doi:10.1063/1.1674756
Return to citation in text: [1] -
Tsumatori, H.; Nakashima, T.; Kawai, T. Org. Lett. 2010, 12, 2362–2365. doi:10.1021/ol100701w
Return to citation in text: [1] -
Schweicher, G.; Lemaur, V.; Niebel, C.; Ruzié, C.; Diao, Y.; Goto, O.; Lee, W.-Y.; Kim, Y.; Arlin, J.-B.; Karpinska, J.; Kennedy, A. R.; Parkin, S. R.; Olivier, Y.; Mannsfeld, S. C. B.; Cornil, J.; Geerts, Y. H.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2015, 27, 3066–3072. doi:10.1002/adma.201500322
Return to citation in text: [1] -
Sugino, H.; Takimiya, K. Chem. Lett. 2017, 46, 345–347. doi:10.1246/cl.161020
Return to citation in text: [1] -
Kohl, B.; Bohnwagner, M. V.; Rominger, F.; Wadepohl, H.; Dreuw, A.; Mastalerz, M. Chem. – Eur. J. 2016, 22, 646–655. doi:10.1002/chem.201503863
Return to citation in text: [1] -
Wu, T.-L.; Kuo, C.-H.; Lin, B.-C.; Tao, Y.-T.; Hsu, C.-P.; Liu, R.-S. J. Mater. Chem. C 2015, 3, 7583–7588. doi:10.1039/c5tc01455g
Return to citation in text: [1] -
Mallory, F. B.; Mallory, C. W.; Regan, C. K.; Aspden, R. J.; Ricks, A. B.; Racowski, J. M.; Nash, A. I.; Gibbons, A. V.; Carroll, P. J.; Bohen, J. M. J. Org. Chem. 2013, 78, 2040–2045. doi:10.1021/jo3020819
Return to citation in text: [1] -
Balaraman, E.; Kumara Swamy, K. C. Tetrahedron: Asymmetry 2007, 18, 2037–2048. doi:10.1016/j.tetasy.2007.06.028
Return to citation in text: [1]
| 49. | Tsumatori, H.; Nakashima, T.; Kawai, T. Org. Lett. 2010, 12, 2362–2365. doi:10.1021/ol100701w |
| 47. | Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. Chem. – Asian J. 2012, 7, 2836–2841. doi:10.1002/asia.201200725 |
| 48. | Emeis, C. A.; Oosterhoff, L. J. J. Chem. Phys. 1971, 54, 4809–4819. doi:10.1063/1.1674756 |
| 1. |
Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567
See for a recent review on the oxidative coupling. |
| 18. | Sánchez-Carnerero, E. M.; Agarrabeitia, A. R.; Moreno, F.; Maroto, B. L.; Muller, G.; Ortiz, M. J.; de la Moya, S. Chem. – Eur. J. 2015, 21, 13488–13500. doi:10.1002/chem.201501178 |
| 19. | Tanaka, H.; Inoue, Y.; Mori, T. ChemPhotoChem 2018, 2, 386–402. doi:10.1002/cptc.201800015 |
| 20. | Chen, N.; Yan, B. Molecules 2018, 23, 3376. doi:10.3390/molecules23123376 |
| 21. | Ma, J.-L.; Peng, Q.; Zhao, C.-H. Chem. – Eur. J. 2019, 25, 15441–15454. doi:10.1002/chem.201903252 |
| 43. | Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t |
| 44. | Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645 |
| 14. | Kaida, H.; Goya, T.; Nishii, Y.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2017, 19, 1236–1239. doi:10.1021/acs.orglett.7b00323 |
| 45. | Sperotto, E.; de Vries, J. G.; van Klink, G. P. M.; van Koten, G. Tetrahedron Lett. 2007, 48, 7366–7370. doi:10.1016/j.tetlet.2007.08.026 |
| 46. | Liu, X.; Zhang, S. Synlett 2011, 268–272. doi:10.1055/s-0030-1259291 |
| 12. | Kaida, H.; Satoh, T.; Hirano, K.; Miura, M. Chem. Lett. 2015, 44, 1125–1127. doi:10.1246/cl.150408 |
| 13. | Kaida, H.; Satoh, T.; Nishii, Y.; Hirano, K.; Miura, M. Chem. Lett. 2016, 45, 1069–1071. doi:10.1246/cl.160496 |
| 14. | Kaida, H.; Goya, T.; Nishii, Y.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2017, 19, 1236–1239. doi:10.1021/acs.orglett.7b00323 |
| 15. | Itai, Y.; Nishii, Y.; Stachelek, P.; Data, P.; Takeda, Y.; Minakata, S.; Miura, M. J. Org. Chem. 2018, 83, 10289–10302. doi:10.1021/acs.joc.8b01451 |
| 16. | Nakamura, S.; Tohnai, N.; Nishii, Y.; Hinoue, T.; Miura, M. ChemPhotoChem 2019, 3, 46–53. doi:10.1002/cptc.201800189 |
| 17. | Nakamura, S.; Okamoto, M.; Tohnai, N.; Nakayama, K.-i.; Nishii, Y.; Miura, M. Bull. Chem. Soc. Jpn. 2020, 93, 99–108. doi:10.1246/bcsj.20190269 |
| 40. | Meca, L.; Řeha, D.; Havlas, Z. J. Org. Chem. 2003, 68, 5677–5680. doi:10.1021/jo034344u |
| 41. | Moustafa, G. A. I.; Oki, Y.; Akai, S. Angew. Chem., Int. Ed. 2018, 57, 10278–10282. doi:10.1002/anie.201804161 |
| 2. | Yoshimoto, H.; Itatani, H. Bull. Chem. Soc. Jpn. 1973, 46, 2490–2492. doi:10.1246/bcsj.46.2490 |
| 3. | Shiotani, A.; Itatani, H. Angew. Chem., Int. Ed. Engl. 1974, 13, 471–472. doi:10.1002/anie.197404711 |
| 4. | Åkermark, B.; Eberson, L.; Jonsson, E.; Pettersson, E. J. Org. Chem. 1975, 40, 1365–1367. doi:10.1021/jo00897a048 |
| 5. | Knölker, H.-J.; O'Sullivan, N. Tetrahedron 1994, 50, 10893–10908. doi:10.1016/s0040-4020(01)85701-x |
| 6. | Åkermark, B.; Oslob, J. D.; Heuschert, U. Tetrahedron Lett. 1995, 36, 1325–1326. doi:10.1016/0040-4039(94)02467-p |
| 7. | Hagelin, H.; Oslob, J. D.; Åkermark, B. Chem. – Eur. J. 1999, 5, 2413–2416. doi:10.1002/(sici)1521-3765(19990802)5:8<2413::aid-chem2413>3.0.co;2-3 |
| 8. | Knölker, H.-J.; Fröhner, W.; Reddy, K. R. Synthesis 2002, 557–564. doi:10.1055/s-2002-20953 |
| 9. | Matsubara, S.; Asano, K.; Kajita, Y.; Yamamoto, M. Synthesis 2007, 2055–2059. doi:10.1055/s-2007-983738 |
| 10. | Watanabe, T.; Ueda, S.; Inuki, S.; Oishi, S.; Fujii, N.; Ohno, H. Chem. Commun. 2007, 4516–4518. doi:10.1039/b707899d |
| 11. | Liégault, B.; Lee, D.; Huestis, M. P.; Stuart, D. R.; Fagnou, K. J. Org. Chem. 2008, 73, 5022–5028. doi:10.1021/jo800596m |
| 42. | Paul, A.; Kumar, A.; Nanjunda, R.; Farahat, A. A.; Boykin, D. W.; Wilson, W. D. Org. Biomol. Chem. 2017, 15, 827–835. doi:10.1039/c6ob02390h |
| 25. | Kawai, T.; Kawamura, K.; Tsumatori, H.; Ishikawa, M.; Naito, M.; Fujiki, M.; Nakashima, T. ChemPhysChem 2007, 8, 1465–1468. doi:10.1002/cphc.200600747 |
| 26. | Amako, T.; Kimoto, T.; Tajima, N.; Fujiki, M.; Imai, Y. Tetrahedron 2013, 69, 2753–2757. doi:10.1016/j.tet.2013.01.084 |
| 27. | Nakabayashi, K.; Amako, T.; Tajima, N.; Fujiki, M.; Imai, Y. Chem. Commun. 2014, 50, 13228–13230. doi:10.1039/c4cc02946a |
| 28. | Feuillastre, S.; Pauton, M.; Gao, L.; Desmarchelier, A.; Riives, A. J.; Prim, D.; Tondelier, D.; Geffroy, B.; Muller, G.; Clavier, G.; Pieters, G. J. Am. Chem. Soc. 2016, 138, 3990–3993. doi:10.1021/jacs.6b00850 |
| 29. | Kitatobe, T.; Mimura, Y.; Tsujimoto, S.; Tajima, N.; Fujiki, M.; Imai, Y. Tetrahedron 2017, 73, 6856–6862. doi:10.1016/j.tet.2017.10.036 |
| 30. | Zhang, X.; Zhang, Y.; Li, Y.; Quan, Y.; Cheng, Y.; Li, Y. Chem. Commun. 2019, 55, 9845–9848. doi:10.1039/c9cc04289j |
| 31. | Zhang, X.; Zhang, Y.; Zhang, H.; Quan, Y.; Li, Y.; Cheng, Y.; Ye, S. Org. Lett. 2019, 21, 439–443. doi:10.1021/acs.orglett.8b03620 |
| 32. | Wu, Z.-G.; Han, H.-B.; Yan, Z.-P.; Luo, X.-F.; Wang, Y.; Zheng, Y.-X.; Zuo, J.-L.; Pan, Y. Adv. Mater. (Weinheim, Ger.) 2019, 31, 1900524. doi:10.1002/adma.201900524 |
| 34. | Takaishi, K.; Yamamoto, T.; Hinoide, S.; Ema, T. Chem. – Eur. J. 2017, 23, 9249–9252. doi:10.1002/chem.201702143 |
| 35. | Takaishi, K.; Takehana, R.; Ema, T. Chem. Commun. 2018, 54, 1449–1452. doi:10.1039/c7cc09187g |
| 36. | Takaishi, K.; Iwachido, K.; Takehana, R.; Uchiyama, M.; Ema, T. J. Am. Chem. Soc. 2019, 141, 6185–6190. doi:10.1021/jacs.9b02582 |
| 37. | Droz, A. S.; Neidlein, U.; Anderson, S.; Seiler, P.; Diederich, F. Helv. Chim. Acta 2001, 84, 2243–2289. doi:10.1002/1522-2675(20010815)84:8<2243::aid-hlca2243>3.0.co;2-g |
| 38. | Ye, P.; Li, Q.; Bai, Z.; Dong, K.; Liu, Q. Heterocycles 2015, 91, 1986–1995. doi:10.3987/com-15-13302 |
| 39. | Octa-Smolin, F.; van der Vight, F.; Yadav, R.; Bhangu, J.; Soloviova, K.; Wölper, C.; Daniliuc, C. G.; Strassert, C. A.; Somnitz, H.; Jansen, G.; Niemeyer, J. J. Org. Chem. 2018, 83, 14568–14587. doi:10.1021/acs.joc.8b02353 |
| 23. | Sherson, J. F.; Krauter, H.; Olsson, R. K.; Julsgaard, B.; Hammerer, K.; Cirac, I.; Polzik, E. S. Nature 2006, 443, 557–560. doi:10.1038/nature05136 |
| 50. | Schweicher, G.; Lemaur, V.; Niebel, C.; Ruzié, C.; Diao, Y.; Goto, O.; Lee, W.-Y.; Kim, Y.; Arlin, J.-B.; Karpinska, J.; Kennedy, A. R.; Parkin, S. R.; Olivier, Y.; Mannsfeld, S. C. B.; Cornil, J.; Geerts, Y. H.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2015, 27, 3066–3072. doi:10.1002/adma.201500322 |
| 51. | Sugino, H.; Takimiya, K. Chem. Lett. 2017, 46, 345–347. doi:10.1246/cl.161020 |
| 52. | Kohl, B.; Bohnwagner, M. V.; Rominger, F.; Wadepohl, H.; Dreuw, A.; Mastalerz, M. Chem. – Eur. J. 2016, 22, 646–655. doi:10.1002/chem.201503863 |
| 53. | Wu, T.-L.; Kuo, C.-H.; Lin, B.-C.; Tao, Y.-T.; Hsu, C.-P.; Liu, R.-S. J. Mater. Chem. C 2015, 3, 7583–7588. doi:10.1039/c5tc01455g |
| 54. | Mallory, F. B.; Mallory, C. W.; Regan, C. K.; Aspden, R. J.; Ricks, A. B.; Racowski, J. M.; Nash, A. I.; Gibbons, A. V.; Carroll, P. J.; Bohen, J. M. J. Org. Chem. 2013, 78, 2040–2045. doi:10.1021/jo3020819 |
| 22. | Schadt, M. Annu. Rev. Mater. Sci. 1997, 27, 305–379. doi:10.1146/annurev.matsci.27.1.305 |
| 33. | Hassan, K.; Yamashita, K.-i.; Hirabayashi, K.; Shimizu, T.; Nakabayashi, K.; Imai, Y.; Matsumoto, T.; Yamano, A.; Sugiura, K.-i. Chem. Lett. 2015, 44, 1607–1609. doi:10.1246/cl.150704 |
| 55. | Balaraman, E.; Kumara Swamy, K. C. Tetrahedron: Asymmetry 2007, 18, 2037–2048. doi:10.1016/j.tetasy.2007.06.028 |
© 2020 Takishima et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)