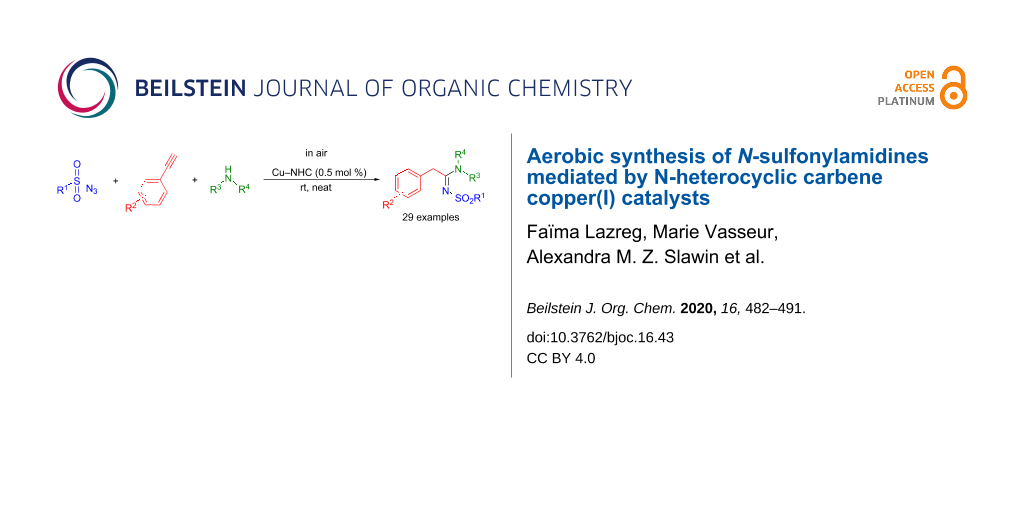
Abstract
A new catalytic strategy for the one-pot synthesis of N-sulfonylamidines is described. The cationic copper(I) complexes were found to be highly active and efficient under mild conditions in air and in the absence of solvent. A copper acetylide is proposed as key intermediate in this transformation.

Graphical Abstract
Introduction
Amide derivatives represent a ubiquitous molecular construct in chemistry [1-3]. This structural motif favours rearrangements that lead to high reactivity and associated bioactivity [4,5]. Indeed, the presence of an N-atom in the amidine structure leads to opportunities as ligands and organocatalysts [6-8]. N-Sulfonylamidines and N-sulfonylimidates are members of a specific class of these amidines. One initial methodology developed for the formation of sulfonylamidines was based on the cleavage of the bond between the N-4 and C-benzene in thiadiazine ring-type molecules [9]. To date, only few examples of copper-based catalysts have been reported to enable access to such compounds [10-13]. Chang and co-workers were pioneers in this area [10-12]. A three-component reaction between alkyne, sulfonyl azide and amine/alcohol was described as a synthetic route to generate sulfonyltriazole intermediates. However, the presence of additives and high catalyst loading (CuI 10 mol %) were required for the synthesis of N-sulfonylimidates (Scheme 1, left).
![[1860-5397-16-43-i1]](/bjoc/content/inline/1860-5397-16-43-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of sulfonyltriazoles and sulfonamidines.
Scheme 1: Formation of sulfonyltriazoles and sulfonamidines.
Over the last two decades, NHCs (NHC = N-heterocyclic carbene) have become ligands of choice to permit the stabilisation and formation of highly reactive transition metal species [14]. Thus, significant advances have been achieved using this supporting ligand family [15-19]. Recently, our group contributed to this area, reporting on the synthesis of the first heteroleptic bis-NHC and mixed NHC/phosphine copper(I) complexes [20-22]. Interestingly, these new copper-based complexes have shown excellent activity in the [3 + 2] cycloaddition reaction of azides/sulfonyl azides and alkynes (Scheme 1, right) [22]. Based on these earlier results, the reactivity of these catalysts was investigated in the context of achieving formation of the challenging sulfonamide derivatives.
Herein, we report the high efficiency of cationic copper(I) complexes for the formation of N-sulfonylamidines via a three-component reaction performed in air, using solvent-free conditions and in the absence of any additive.
Results and Discussion
[Cu(ICy)2]BF4 (1), [Cu(IPr)(ICy)]BF4 (2) and [Cu(IPr)(Pt-Bu3)]BF4 (3) were initially selected as optimum candidates [22]. This class of catalysts was expanded through the synthesis of the pyridine derivative 4, of the heteroleptic normal/mesoionic carbene complex 5, and of the homoleptic mesoionic triazole derivative 6 (Figure 1). This special class of ligands presents unique electronic and steric properties and lead to unusual reactivity [23-28].
![[1860-5397-16-43-1]](/bjoc/content/figures/1860-5397-16-43-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Catalytic systems used in this study.
Figure 1: Catalytic systems used in this study.
[Cu(IPr)(Pyr)]OTf (4) was obtained by the reaction of the isolated hydroxide derivative [Cu(IPr)(OH)] [29] with pyridinium trifluoromethanesulfonate, while the biscarbene complexes 5 and 6 were obtained from the corresponding [Cu(NHC)Cl] through the in situ formation of the corresponding hydroxide complex [Cu(NHC)(OH)] [20] which deprotonates the triazolium salt (Scheme 2).
![[1860-5397-16-43-i2]](/bjoc/content/inline/1860-5397-16-43-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic access to complexes 4–6 [30].
Scheme 2: Synthetic access to complexes 4–6 [30].
The reactivity of a series of cationic copper(I) complexes (1–6) was evaluated at 0.5 mol % loading using tosyl azide, phenylacetylene and diisopropylamine as benchmark substrates [31,32]. Various solvents were evaluated at room temperature under aerobic conditions (see Supporting Information File 1 for details). Tetrahydrofuran (THF), 1,2-DCE, 1,4-dioxane and acetonitrile proved to be the most suitable solvents for this transformation (Table 1). Interestingly, similar results were obtained for complexes 1–5, while 6 displayed superior activity. Indeed, 71% conversion to the desired product was observed using the homoleptic cationic MIC (mesoionic carbene) complex 6 (Table 1, entry 6). For complexes 1–5, the conversion proved modest and ranged between 42 and 49% (Table 1, entries 1–5).
Table 1: Catalyst and solvent optimisation.a,b
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-43-i12.svg?max-width=637&scale=1.0)
|
|||
| Entry | Complex | Solvent | Conv.c (%) |
| 1 | 1 | THF | 49 |
| 2 | 2 | THF | 47 |
| 3 | 3 | THF | 42 |
| 4 | 4 | THF | 42 |
| 5 | 5 | THF | 46 |
| 6 | 6 | THF | 71 |
| 7 | 6 | neat | 65 |
| 8 | 6 | 1,2-DCE | 63 |
| 9 | 6 | water | 41 |
| 10 | 6 | 1,4-dioxane | 58 |
| 11 | 1 | neat | 30 |
| 12 | 2 | neat | 58 |
| 13 | 5 | neat | 55 |
aReaction conditions: phenylacetylene (0.5 mmol), tosyl azide (0.6 mmol), diisopropylamine (0.6 mmol), [Cu] (0.5 mol %), solvent (1 mL), 16 hours. bSee Supporting Information File 1 for full optimisation. cConversion was determined by GC analysis based on phenylacetylene using mesitylene (42 µL) as internal standard.
Subsequently, solvent-free conditions were investigated (Table 1, entries 7 and 11–13). Interestingly, the absence of solvent proved to be highly effective, except for [Cu(ICy)2]BF4 1 (Table 1 entry 11). An encouraging 65% conversion was obtained using [Cu(Triaz)2]BF4 6 (Table 1, entry 7), while complexes 2 and 5 showed comparable results (Table 1, entries 12 and 13). Complex 6 was also shown to be active in water and in 1,4-dioxane. Based on these results, a reaction scope was conducted under solvent-free conditions, in air, using 1 mol % of [Cu(Triaz)2]BF4 6 (Scheme 3).
![[1860-5397-16-43-i3]](/bjoc/content/inline/1860-5397-16-43-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Variation of sulfonylazides. Reaction conditions: phenylacetylene (0.5 mmol), sulfonyl azide (0.6 mmol), diisopropylamine (0.6 mmol), 6 (1 mol %). Conversion determined by 1H NMR based on alkyne using mesitylene (42 µL) as internal standard. Isolated yield in parentheses. aSolvent-free conditions, rt. bSolvent-free conditions, 40 °C. cTHF (1 mL), rt.
Scheme 3: Variation of sulfonylazides. Reaction conditions: phenylacetylene (0.5 mmol), sulfonyl azide (0.6 m...
Functionalised azides were reacted with phenylacetylene and diisopropylamine resulting in good to high yields (Scheme 3, 10a–g). The presence of an activating/deactivating group in para-position of the aryl ring was evaluated in order to assess the substrate tolerance. Electron-donating groups, such as methyl, methoxy or naphthyl enhanced considerably the reactivity leading to quantitative conversion with respectively 96% (10a), 95% (10c) and 93% (10g) isolated yields, in reaction times of 3 to 4.5 hours. Regarding the 2,4,6-triisopropylsulfonyl azide, a slight decrease in the reactivity was observed (66%, 10f), presumably due to the steric hindrance of the substrate. Electron-withdrawing groups such as bromo (10d) or cyano (10e) appeared to disfavour the reaction resulting in lower yields. Indeed for the cyanosulfonyl azide, under solvent-free conditions, only 38% of the desired product was obtained after 24 hours. This lower yield could also be due to the starting material being a solid which leads to poorer mixing and mass transport issues. Interestingly, when conducted in THF, an increase to 68% isolated yield was observed after 4 hours, supporting our inhomogeneity/transport hypothesis. In the case of the bromosulfonyl azide, a 73% conversion was obtained after 4 hours at 40 °C.
Various terminal aryl/alkyl-substituted alkynes were investigated in the presence of tosyl azide and diisopropylamine (Scheme 4). Under standard conditions, good to excellent yields were obtained. The presence of functional groups in para-position of the aryl ring leads to a decrease of the conversion to approximately 50% (11a, 11b, 11c and 11d). The reactivity was considerably enhanced by increasing the temperature to 40 °C and/or the use of THF as reaction solvent. Interestingly, ortho- and meta-substitution of the aryl rings are well tolerated (11e–i). In the case of 9-ethynylphenanthrene, which is a solid substrate, solvent-free conditions lead to 78% isolated yield at 40 °C. Diynes were also investigated and excellent isolated yields were achieved (92% and 85% for 11j and 11k, respectively). Regarding the alkyl-alkynes, longer reactions times as well as higher temperature were required to reach high conversion. Interestingly, in the case of 11r, the desired product was obtained in 67% isolated yield.
![[1860-5397-16-43-i4]](/bjoc/content/inline/1860-5397-16-43-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Variation of alkynes. Reaction conditions: alkyne (0.5 mmol), tosyl azide (0.6 mmol), diisopropylamine (0.6 mmol), 6 (1 mol %). Conversion determined by 1H NMR based on alkyne using mesitylene (42 µL) as internal standard. Isolated yield in parenthesis. aSolvent-free conditions, rt. bSolvent-free conditions, 40 °C. cTHF (1 mL), rt.
Scheme 4: Variation of alkynes. Reaction conditions: alkyne (0.5 mmol), tosyl azide (0.6 mmol), diisopropylam...
The effect of the amines was also investigated. Amongst the amines evaluated, dicyclohexylamine (for 12a) and isopropylamine (for 12b) lead to good isolated yields (64% and 72%, Scheme 5). In contrast, with diphenylamine, only 20% of the desired product was observed (12c).
![[1860-5397-16-43-i5]](/bjoc/content/inline/1860-5397-16-43-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Variation of the amine substrate. Reaction conditions: phenylacetylene (0.5 mmol), tosyl azide (0.6 mmol), amine (0.6 mmol), 6 (1 mol %). Conversion determined by 1H NMR based on alkyne using mesitylene (42 µL) as internal standard. Isolated yield in parenthesis. aSolvent-free conditions, 40 °C. bTHF (1 mL), rt.
Scheme 5: Variation of the amine substrate. Reaction conditions: phenylacetylene (0.5 mmol), tosyl azide (0.6...
Interestingly, with benzyl azide, a substrate not containing a sulfonyl moiety, the product obtained is the 1,2,3-triazole derivative [33], resulting from a [3 + 2] cycloaddition of azide and alkyne (Scheme 6).
![[1860-5397-16-43-i6]](/bjoc/content/inline/1860-5397-16-43-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactivity of “non-sulfonyl” azide [33]. Reaction conditions: phenylacetylene (0.5 mmol), benzyl azide (0.6 mmol), diisopropylamine (0.6 mmol), 24 h.
Scheme 6: Reactivity of “non-sulfonyl” azide [33]. Reaction conditions: phenylacetylene (0.5 mmol), benzyl azide ...
The catalytic system was also shown applicable to phosphoryl azides; and reaction of phenylacetylene with diisopropylamine and diphenylphosphoryl azide leads to the formation of the corresponding phosphorylamidine product [34] in good yield (Scheme 7), using 2 mol % of catalyst under mild conditions (solvent-free, room temperature).
![[1860-5397-16-43-i7]](/bjoc/content/inline/1860-5397-16-43-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reactivity of diphenylphosphoryl azide. Reaction conditions: phenylacetylene (0.5 mmol), diphenylphosphoryl azide (0.6 mmol), diisopropylamine (0.6 mmol), [Cu] (2 mol %), 24 h. Conversion determined by GC based on phenylacetylene using mesitylene (42 µL) as internal standard. Isolated yield in parentheses.
Scheme 7: Reactivity of diphenylphosphoryl azide. Reaction conditions: phenylacetylene (0.5 mmol), diphenylph...
A proposed reaction mechanism occurring via formation of a copper-acetylide species is proposed and illustrated in Scheme 8. The bis-NHC copper(I) complex 6 reacts with the alkyne leading to the formation of an acetylide derivative A (left hand side, Scheme 8), with concomitant loss of a NHC ligand through the formation of the corresponding triazolium salt B. The intermediate A can then react with the azide substrate to form a triazolyl–copper complex C. The latter can liberate the amidine product and regenerate either catalyst 6 (triazolium salt B is source of proton) or directly the acetylide complex A (phenylacetylene is source of proton).
![[1860-5397-16-43-i8]](/bjoc/content/inline/1860-5397-16-43-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Proposed mechanism for the formation of sulfonamidine.
Scheme 8: Proposed mechanism for the formation of sulfonamidine.
In order to support this mechanism, a number of stoichiometric reactions were conducted (see Supporting Information File 1). In a first instance, 6 was reacted with phenylacetylene at room temperature. This led to the rapid formation of the copper acetylide complex A with concomitant loss of the triazolium salt Triaz.HBF4 (B, Scheme 9). To further confirm the formation of A, [Cu(Triaz)Cl] was reacted with phenylacetylene and sodium hydroxide (4 equiv), in toluene for 24 hours under an inert atmosphere. The independent synthesis of A was successfully achieved in this manner (Scheme 10). The latter was then reacted with tosyl azide. An immediate colour change resulted and based on 1H NMR data, two new species were formed. They were identified as the sulfonyltriazole and an unstable organometallic compound, presumably the triazolylcopper(I) complex C. These results corroborated the proposed hypothesis regarding the formation of a triazole intermediate during the catalytic cycle.
![[1860-5397-16-43-i9]](/bjoc/content/inline/1860-5397-16-43-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Stoichiometric reaction between 6 and 8.
Scheme 9: Stoichiometric reaction between 6 and 8.
![[1860-5397-16-43-i10]](/bjoc/content/inline/1860-5397-16-43-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of copper-acetylide intermediate A via [Cu(Cl)(Triaz)].
Scheme 10: Synthesis of copper-acetylide intermediate A via [Cu(Cl)(Triaz)].
To support the relevance of the Cu-acetylide species in catalysis, the benchmark reaction was conducted at 1 mol % catalyst of the isolated acetylide complex (Scheme 11). After 45 minutes, 80% conversion into the sulfonamidine product was observed. Of note, the presence of sulfonyltriazole was also observed (10%).
![[1860-5397-16-43-i11]](/bjoc/content/inline/1860-5397-16-43-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Catalytic reaction involving copper-acetylide complex A.
Scheme 11: Catalytic reaction involving copper-acetylide complex A.
Conclusion
Cationic bis-carbene copper(I) complexes were shown to promote the formation of N-sulfonamidines in a Click reaction [35,36]. The new developed mesoionic NHC copper(I) complexes were found highly efficient under solvent-free and aerobic conditions. Stoichiometric reactions support the release of one NHC and the formation of a copper(I) acetylide as key elements in the catalytic cycle.
Experimental
N,N’-Bis{2,6-(diisopropyl)phenyl}imidazol-2-ylidene(pyridine)copper(I) triflate, [Cu(IPr)(Pyr)]OTf (4). In a glovebox, a vial was charged with [Cu(OH)(IPr)] (200 mg, 0.41 mmol), pyridinium trifluoromethanesulfonate (94.0 mg, 1 equiv, 0.41 mmol) and THF (2 mL). The reaction mixture was stirred at room temperature for 15 hours. The solution was concentrated and diethyl ether (10 mL) was added. The precipitate was collected by filtration and washed with diethyl ether (3 × 5 mL). The desired compound was obtained as a colourless solid (201 mg, 92%). 1H NMR (400 MHz, CD2Cl2, 298 K) δ (ppm) 0.82 (d, 3JHH = 6.9 Hz, 12H, CHCH3 (IPr)), 1.01 (d, 3JHH = 6.9 Hz, 12H, CHCH3 (IPr)), 1.44 (s, 9H, C(CH3)3), 2.36 (septet, 3JHH = 6.9 Hz, 4H, CHCH3 (IPr)), 3.98 (s, 3H, CH3), 7.06 (s, 2H, H4 and H5), 7.19 (d, 3JHH = 7.8 Hz, 4H, CArH (IPr)), 7.50 (t, 3JHH = 7.8 Hz, 2H, CArH (IPr)); 13C{1H} NMR (100 MHz, CD2Cl2, 298 K, TMS) δ (ppm) 23.5 (s, CHCH3 (Triaz)), 24.8 (s, CHCH3 (Triaz)), 28.8 (s, 2 CHCH3 (Triaz)), 124.4 (s, CArH), 124.5 (s, CArH), 131.0 (s, CArH), 134.2 (s, CArH), 140.8 (s, CIV), 145.8 (s, CH (IPr)), 148.9 (s, CIV), 177.5 (s, Ccarbene); 19F{1H} NMR (282 MHz, CDCl3, 298 K) δ (ppm) −78.6 (s); anal. calcd for C33H41CuF3N3O3S: C, 58.26; H, 6.07; N, 6.18; found: C, 58.39; H, 6.16; N, 6.08.
1-{2,6-(Diisopropyl)phenyl}-3-methyl-4-(4-tert-butylphenyl)-1,2,3-triazol-5-ylidene-(N,N’-bis{2,6-(diisopropyl)phenyl}imidazol-2-ylidene)copper(I) tetrafluoroborate, [Cu(IPr)(Triaz)]BF4 (5). In a glovebox, a microwave vial was charged with [Cu(Cl)(IPr)] (200.0 mg, 0.41 mmol), NaOH (66.0 mg, 4 equiv, 1.64 mmol), Triaz.HBF4 (190.0 mg, 1 equiv, 0.41 mmol) and acetonitrile (2 mL). The reaction mixture was stirred during 2 h at 80 °C in a microwave. The solution was concentrated and diethyl ether (10 mL) was added. The precipitate was collected by filtration and washed with diethyl ether (3 × 5 mL). The desired compound was obtained as a colourless solid (346 mg, 92%). 1H NMR (400 MHz, CD2Cl2, 298 K) δ (ppm) 0.77 (d, 3JHH = 6.9 Hz, 6H, CHCH3 (Triaz)), 0.82 (d, 3JHH = 6.9 Hz, 12H, CHCH3 (IPr)), 0.96 (d, 3JHH = 6.9 Hz, 6H, CHCH3 (Triaz)), 1.01 (d, 3JHH = 6.9 Hz, 12H, CHCH3 (IPr)), 1.44 (s, 9H, C(CH3)3) 1.99 (septet, 3JHH = 6.9 Hz, 2H, CHCH3 (Triaz)), 2.36 (septet, 3JHH = 6.9 Hz, 4H, CHCH3 (IPr)), 3.98 (s, 3H, CH3), 6.98 (d, 3JHH = 8.3 Hz, 2H, CArH (Triaz)), 7.06 (s, 2H, H4 and H5), 7.10 (d, 3JHH = 7.9 Hz, 2H, CArH (Triaz)), 7.19 (d, 3JHH = 7.8 Hz, 4H, CArH (IPr)), 7.36 (d, 3JHH = 8.4 Hz, 2H, CArH (Triaz)), 7.43 (t, 3JHH = 7.9 Hz, 1H, CArH (Triaz)), 7.50 (t, 3JHH = 7.8 Hz, 2H, CArH (IPr)); 13C{1H} NMR (75 MHz, CD2Cl2, 298 K) δ (ppm) 23.4 (s, CHCH3 (Triaz)), 24.8 (s, CHCH3 (IPr)), 24.4 (s, CHCH3 (IPr)), 24.7 (s, CHCH3 (Triaz)), 28.4 (s, CHCH3 (Triaz)), 28.6 (s, CHCH3 (IPr)), 31.3 (s, CCH3), 35.0 (s, CIV), 37.6 (s, CH3), 123.4 (s, CIV), 123.9 (s, CArH), 124.2 (s, C4 and C5), 124.4 (s, CArH), 126.6 (s, CArH), 129.4 (s, CArH), 130.5 (s, CArH), 130.9 (s, CArH), 134.3 (s, CIV), 144.8 (s, CIV), 145.2 (s, CIV), 152.0 (s, CIV), 152.8 (s, CIV), 179.4 (s, Ccarbene); 19F{1H} NMR (282 MHz, CDCl3, 298 K) δ (ppm) −155.0 (s, BF4), −155.1 (s, BF4); anal. calcd for C52H69BCuF4N5: C, 68.30; H, 7.61; N, 7.66; found: C, 68.15; H, 7.72; N, 7.68.
Bis{1-{2,6-(diisopropyl)phenyl}-3-methyl-4-(4-tert-butylphenyl)-1,2,3-triazol-5-ylidene}copper(I) tetrafluoroborate, [Cu(Triaz)2]BF4 (6). In a glovebox, a vial was charged with [Cu(Cl)(Triaz)] (150.0 mg, 0.32 mmol), NaOH (50 mg, 4 equiv, 1.28 mmol), Triaz.HBF4 (148 mg, 1 equiv, 0.32 mmol) and acetonitrile (2 mL). The reaction mixture was stirred during 2 h at 80 °C in a microwave. The solution was concentrated (0.5 mL) and diethyl ether (10 mL) was added. The precipitate was collected by filtration and washed with diethyl ether (3 × 5 mL). The desired compound was obtained as a colourless solid (281 mg, 97%). 1H NMR (400 MHz, CDCl3, 298 K, TMS) δ (ppm) 0.79 (d, 3JHH = 6.8 Hz, 12H, CHCH3 (IPr)), 1.08 (d, 3JHH = 6.8 Hz, 12H, CHCH3 (IPr)), 1.38 (s, 18H, C(CH3)3), 2.10 (septet, 3JHH = 6.8 Hz, 4H, CHCH3 (IPr)), 4.20 (s, 6H, CH3), 7.19 (d, 3JHH = 7.8 Hz, 4H, CArH (IPr)), 7.34 (m, 8H, CArH), 7.49 (t, 3JHH = 7.7 Hz, 2H, CArH (IPr)); 13C{1H} NMR (75 MHz, CDCl3, 298 K, TMS) δ (ppm) 23.8 (s, CHCH3 (Triaz)), 24.2 (s, CHCH3 (Triaz)), 28.4 (s, CHCH3 (Triaz)), 31.3 (s, CCH3), 35.0 (s, CIV), 37.8 (s, CH3), 123.6 (s, CIV), 124 (s, CArH), 126.2 (s, CArH), 129.0 (s, CArH), 131.1 (s, CArH), 134.3 (s, CIV), 145.0 (s, CIV), 149.2 (s, CIV), 153.4 (s, CIV); 19F{1H} NMR (282 MHz, CDCl3, 298 K) δ (ppm) −154.9 (s, BF4), −155.0 (s, BF4); anal. calcd for C50H66BCuF4N6: C, 66.62; H, 7.38; N, 9.32; found: C, 66.55; H, 7.46; N, 9.47.
General catalytic procedure. A vial was charged with [Cu(Triaz)2]BF4 (4.5 mg, 1 mol %), the alkyne (0.5 mmol), the azide (0.6 mmol) and the amine (0.6 mmol). The reaction was stirred neat for the appropriate amount of time. Dichloromethane (2 mL) and a saturated aqueous solution of ammonium chloride (3 mL) were added and the reaction mixture stirred during 30 minutes. The aqueous layer was extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under vacuum. The crude product was purified by flash column chromatography or by recrystallization. The reported yields are the average of two reactions.
Supporting Information
| Supporting Information File 1: Experimental and characterisation data. | ||
| Format: PDF | Size: 4.0 MB | Download |
| Supporting Information File 2: Crystal data for 4. | ||
| Format: CIF | Size: 1.0 MB | Download |
| Supporting Information File 3: Crystal data for 5. | ||
| Format: CIF | Size: 603.3 KB | Download |
| Supporting Information File 4: Crystal data for 6. | ||
| Format: CIF | Size: 1.4 MB | Download |
References
-
Zabicky, J. The Chemistry of Amides; Interscience: London, New York, 1970. doi:10.1002/9780470771235
Return to citation in text: [1] -
Gautier, J.-A.; Miocque, M.; Farnoux, C. C. Preparation and synthetic uses of amidines. Amidines and Imidates Vol. 1 (1975); John Wiley & Sons, Ltd: Chichester, United Kingdom; pp 283–348. doi:10.1002/9780470771495.ch7
Return to citation in text: [1] -
Bielawski, K.; Bielawska, A.; Sosnowska, K.; Miltyk, W.; Winnicka, K.; Pałka, J. Biochem. Pharmacol. 2006, 72, 320–331. doi:10.1016/j.bcp.2006.04.028
Return to citation in text: [1] -
Sondhi, S. M.; Singh, J.; Kumar, A.; Jamal, H.; Gupta, P. P. Eur. J. Med. Chem. 2009, 44, 1010–1015. doi:10.1016/j.ejmech.2008.06.029
Return to citation in text: [1] -
Özden, S.; Atabey, D.; Yıldız, S.; Göker, H. Bioorg. Med. Chem. 2005, 13, 1587–1597. doi:10.1016/j.bmc.2004.12.025
Return to citation in text: [1] -
Barker, J.; Kilner, M. Coord. Chem. Rev. 1994, 133, 219–300. doi:10.1016/0010-8545(94)80059-6
Return to citation in text: [1] -
Oakley, S. H.; Soria, D. B.; Coles, M. P.; Hitchcock, P. B. Dalton Trans. 2004, 537–546. doi:10.1039/b314707j
Return to citation in text: [1] -
Ahmad, S. M.; Braddock, D. C.; Cansell, G.; Hermitage, S. A.; Redmond, J. M.; White, A. J. P. Tetrahedron Lett. 2007, 48, 5948–5952. doi:10.1016/j.tetlet.2007.06.112
Return to citation in text: [1] -
Iwakawa, T.; Tamura, H.; Masuko, M.; Murabayashi, A.; Hayase, Y. J. Pestic. Sci. (Jpn. Ed., 1976-2002) 1992, 17, 131–135. doi:10.1584/jpestics.17.2_131
Return to citation in text: [1] -
Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038–2039. doi:10.1021/ja0432968
Return to citation in text: [1] [2] -
Yoo, E. J.; Ahlquist, M.; Bae, I.; Sharpless, K. B.; Fokin, V. V.; Chang, S. J. Org. Chem. 2008, 73, 5520–5528. doi:10.1021/jo800733p
Return to citation in text: [1] [2] -
Yavari, I.; Ahmadian, S.; Ghazanfarpur-Darjani, M.; Solgi, Y. Tetrahedron Lett. 2011, 52, 668–670. doi:10.1016/j.tetlet.2010.11.135
Return to citation in text: [1] [2] -
Yang, T.; Cui, H.; Zhang, C.; Zhang, L.; Su, C.-Y. Inorg. Chem. 2013, 52, 9053–9059. doi:10.1021/ic4012229
Return to citation in text: [1] -
Shi, S.; Nolan, S. P.; Szostak, M. Acc. Chem. Res. 2018, 51, 2589–2599. doi:10.1021/acs.accounts.8b00410
Return to citation in text: [1] -
Cazin, C. S. J., Ed. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis; Springer: Dordrecht, Netherlands, 2011; Vol. 32. doi:10.1007/978-90-481-2866-2
Return to citation in text: [1] -
Nolan, S. P. N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/9783527609451
Return to citation in text: [1] -
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m
Return to citation in text: [1] -
Raubenheimer, H. G.; Cronje, S.; Olivier, P. J. J. Chem. Soc., Dalton Trans. 1995, 313–316. doi:10.1039/dt9950000313
Return to citation in text: [1] -
Egbert, J. D.; Cazin, C. S. J.; Nolan, S. P. Catal. Sci. Technol. 2013, 3, 912–926. doi:10.1039/c2cy20816d
Return to citation in text: [1] -
Lazreg, F.; Cordes, D. B.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2015, 34, 419–425. doi:10.1021/om500882t
Return to citation in text: [1] [2] -
Lazreg, F.; Cazin, C. J. S. NHC–Copper Complexes and their Applications. N-Heterocyclic Carbenes; Wiley-VCH: Weinheim, Germany, 2014; pp 199–242. doi:10.1002/9783527671229.ch08
Return to citation in text: [1] -
Lazreg, F.; Cazin, C. S. J. Organometallics 2018, 37, 679–683. doi:10.1021/acs.organomet.7b00506
Return to citation in text: [1] [2] [3] -
Yoo, E. J.; Bae, I.; Cho, S. H.; Han, H.; Chang, S. Org. Lett. 2006, 8, 1347–1350. doi:10.1021/ol060056j
Return to citation in text: [1] -
Martin, D.; Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Organometallics 2011, 30, 5304–5313. doi:10.1021/om200650x
Return to citation in text: [1] -
Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s
Return to citation in text: [1] -
Schuster, O.; Yang, L.; Raubenheimer, H. G.; Albrecht, M. Chem. Rev. 2009, 109, 3445–3478. doi:10.1021/cr8005087
Return to citation in text: [1] -
Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2018, 51, 3236–3244. doi:10.1021/acs.accounts.8b00480
Return to citation in text: [1] -
Bidal, Y. D.; Lesieur, M.; Melaimi, M.; Nahra, F.; Cordes, D. B.; Athukorala Arachchige, K. S.; Slawin, A. M. Z.; Bertrand, G.; Cazin, C. S. J. Adv. Synth. Catal. 2015, 357, 3155–3161. doi:10.1002/adsc.201500453
Return to citation in text: [1] -
Fortman, G. C.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2010, 29, 3966–3972. doi:10.1021/om100733n
Return to citation in text: [1] -
CCDC 1443106 (4), CCDC 1443107 (5), CCDC 1443108 (6) contain the supplementary crystallographic data for this contribution. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Shojaei, S.; Ghasemi, Z.; Shahrisa, A. Appl. Organomet. Chem. 2017, 31, e3788. doi:10.1002/aoc.3788
Return to citation in text: [1] -
Mandal, S.; Gauniyal, H. M.; Pramanik, K.; Mukhopadhyay, B. J. Org. Chem. 2007, 72, 9753–9756. doi:10.1021/jo701565m
Return to citation in text: [1] -
Lazreg, F.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2012, 31, 7969–7975. doi:10.1021/om3006195
Return to citation in text: [1] [2] -
Kim, S. H.; Jung, D. Y.; Chang, S. J. Org. Chem. 2007, 72, 9769–9771. doi:10.1021/jo7016247
Return to citation in text: [1] -
Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506
Return to citation in text: [1] -
Jin, L.; Tolentino, D. R.; Melaimi, M.; Bertrand, G. Sci. Adv. 2015, 1, e1500304. doi:10.1126/sciadv.1500304
Return to citation in text: [1]
| 34. | Kim, S. H.; Jung, D. Y.; Chang, S. J. Org. Chem. 2007, 72, 9769–9771. doi:10.1021/jo7016247 |
| 33. | Lazreg, F.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2012, 31, 7969–7975. doi:10.1021/om3006195 |
| 33. | Lazreg, F.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2012, 31, 7969–7975. doi:10.1021/om3006195 |
| 1. | Zabicky, J. The Chemistry of Amides; Interscience: London, New York, 1970. doi:10.1002/9780470771235 |
| 2. | Gautier, J.-A.; Miocque, M.; Farnoux, C. C. Preparation and synthetic uses of amidines. Amidines and Imidates Vol. 1 (1975); John Wiley & Sons, Ltd: Chichester, United Kingdom; pp 283–348. doi:10.1002/9780470771495.ch7 |
| 3. | Bielawski, K.; Bielawska, A.; Sosnowska, K.; Miltyk, W.; Winnicka, K.; Pałka, J. Biochem. Pharmacol. 2006, 72, 320–331. doi:10.1016/j.bcp.2006.04.028 |
| 10. | Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038–2039. doi:10.1021/ja0432968 |
| 11. | Yoo, E. J.; Ahlquist, M.; Bae, I.; Sharpless, K. B.; Fokin, V. V.; Chang, S. J. Org. Chem. 2008, 73, 5520–5528. doi:10.1021/jo800733p |
| 12. | Yavari, I.; Ahmadian, S.; Ghazanfarpur-Darjani, M.; Solgi, Y. Tetrahedron Lett. 2011, 52, 668–670. doi:10.1016/j.tetlet.2010.11.135 |
| 13. | Yang, T.; Cui, H.; Zhang, C.; Zhang, L.; Su, C.-Y. Inorg. Chem. 2013, 52, 9053–9059. doi:10.1021/ic4012229 |
| 30. | CCDC 1443106 (4), CCDC 1443107 (5), CCDC 1443108 (6) contain the supplementary crystallographic data for this contribution. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 9. | Iwakawa, T.; Tamura, H.; Masuko, M.; Murabayashi, A.; Hayase, Y. J. Pestic. Sci. (Jpn. Ed., 1976-2002) 1992, 17, 131–135. doi:10.1584/jpestics.17.2_131 |
| 31. | Shojaei, S.; Ghasemi, Z.; Shahrisa, A. Appl. Organomet. Chem. 2017, 31, e3788. doi:10.1002/aoc.3788 |
| 32. | Mandal, S.; Gauniyal, H. M.; Pramanik, K.; Mukhopadhyay, B. J. Org. Chem. 2007, 72, 9753–9756. doi:10.1021/jo701565m |
| 6. | Barker, J.; Kilner, M. Coord. Chem. Rev. 1994, 133, 219–300. doi:10.1016/0010-8545(94)80059-6 |
| 7. | Oakley, S. H.; Soria, D. B.; Coles, M. P.; Hitchcock, P. B. Dalton Trans. 2004, 537–546. doi:10.1039/b314707j |
| 8. | Ahmad, S. M.; Braddock, D. C.; Cansell, G.; Hermitage, S. A.; Redmond, J. M.; White, A. J. P. Tetrahedron Lett. 2007, 48, 5948–5952. doi:10.1016/j.tetlet.2007.06.112 |
| 29. | Fortman, G. C.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2010, 29, 3966–3972. doi:10.1021/om100733n |
| 4. | Sondhi, S. M.; Singh, J.; Kumar, A.; Jamal, H.; Gupta, P. P. Eur. J. Med. Chem. 2009, 44, 1010–1015. doi:10.1016/j.ejmech.2008.06.029 |
| 5. | Özden, S.; Atabey, D.; Yıldız, S.; Göker, H. Bioorg. Med. Chem. 2005, 13, 1587–1597. doi:10.1016/j.bmc.2004.12.025 |
| 20. | Lazreg, F.; Cordes, D. B.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2015, 34, 419–425. doi:10.1021/om500882t |
| 20. | Lazreg, F.; Cordes, D. B.; Slawin, A. M. Z.; Cazin, C. S. J. Organometallics 2015, 34, 419–425. doi:10.1021/om500882t |
| 21. | Lazreg, F.; Cazin, C. J. S. NHC–Copper Complexes and their Applications. N-Heterocyclic Carbenes; Wiley-VCH: Weinheim, Germany, 2014; pp 199–242. doi:10.1002/9783527671229.ch08 |
| 22. | Lazreg, F.; Cazin, C. S. J. Organometallics 2018, 37, 679–683. doi:10.1021/acs.organomet.7b00506 |
| 22. | Lazreg, F.; Cazin, C. S. J. Organometallics 2018, 37, 679–683. doi:10.1021/acs.organomet.7b00506 |
| 15. | Cazin, C. S. J., Ed. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis; Springer: Dordrecht, Netherlands, 2011; Vol. 32. doi:10.1007/978-90-481-2866-2 |
| 16. | Nolan, S. P. N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. doi:10.1002/9783527609451 |
| 17. | Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/cr900074m |
| 18. | Raubenheimer, H. G.; Cronje, S.; Olivier, P. J. J. Chem. Soc., Dalton Trans. 1995, 313–316. doi:10.1039/dt9950000313 |
| 19. | Egbert, J. D.; Cazin, C. S. J.; Nolan, S. P. Catal. Sci. Technol. 2013, 3, 912–926. doi:10.1039/c2cy20816d |
| 23. | Yoo, E. J.; Bae, I.; Cho, S. H.; Han, H.; Chang, S. Org. Lett. 2006, 8, 1347–1350. doi:10.1021/ol060056j |
| 24. | Martin, D.; Melaimi, M.; Soleilhavoup, M.; Bertrand, G. Organometallics 2011, 30, 5304–5313. doi:10.1021/om200650x |
| 25. | Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s |
| 26. | Schuster, O.; Yang, L.; Raubenheimer, H. G.; Albrecht, M. Chem. Rev. 2009, 109, 3445–3478. doi:10.1021/cr8005087 |
| 27. | Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. Acc. Chem. Res. 2018, 51, 3236–3244. doi:10.1021/acs.accounts.8b00480 |
| 28. | Bidal, Y. D.; Lesieur, M.; Melaimi, M.; Nahra, F.; Cordes, D. B.; Athukorala Arachchige, K. S.; Slawin, A. M. Z.; Bertrand, G.; Cazin, C. S. J. Adv. Synth. Catal. 2015, 357, 3155–3161. doi:10.1002/adsc.201500453 |
| 14. | Shi, S.; Nolan, S. P.; Szostak, M. Acc. Chem. Res. 2018, 51, 2589–2599. doi:10.1021/acs.accounts.8b00410 |
| 35. | Worrell, B. T.; Malik, J. A.; Fokin, V. V. Science 2013, 340, 457–460. doi:10.1126/science.1229506 |
| 36. | Jin, L.; Tolentino, D. R.; Melaimi, M.; Bertrand, G. Sci. Adv. 2015, 1, e1500304. doi:10.1126/sciadv.1500304 |
| 10. | Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038–2039. doi:10.1021/ja0432968 |
| 11. | Yoo, E. J.; Ahlquist, M.; Bae, I.; Sharpless, K. B.; Fokin, V. V.; Chang, S. J. Org. Chem. 2008, 73, 5520–5528. doi:10.1021/jo800733p |
| 12. | Yavari, I.; Ahmadian, S.; Ghazanfarpur-Darjani, M.; Solgi, Y. Tetrahedron Lett. 2011, 52, 668–670. doi:10.1016/j.tetlet.2010.11.135 |
| 22. | Lazreg, F.; Cazin, C. S. J. Organometallics 2018, 37, 679–683. doi:10.1021/acs.organomet.7b00506 |
© 2020 Lazreg et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)