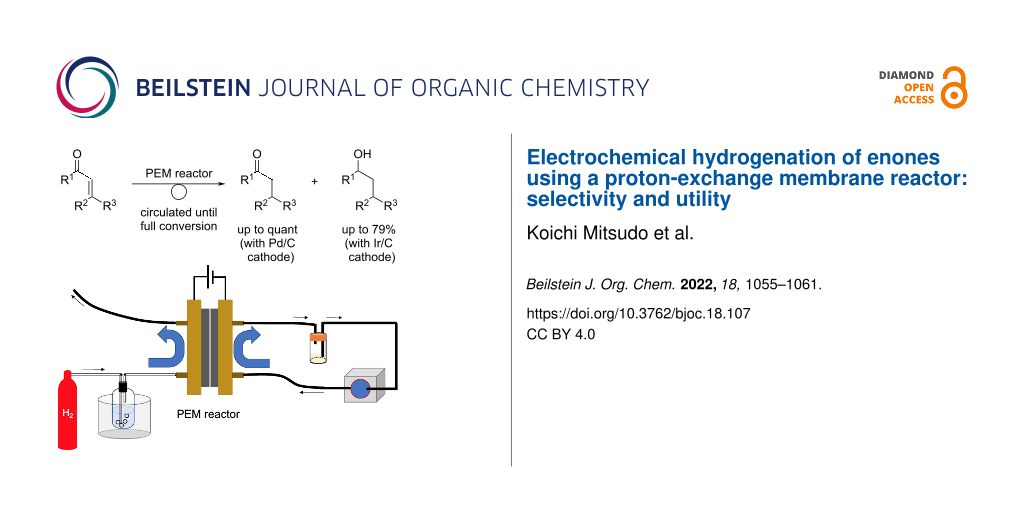
Abstract
Electrochemical hydrogenation of enones using a proton-exchange membrane reactor is described. The reduction of enones proceeded smoothly under mild conditions to afford ketones or alcohols. The reaction occurred chemoselectively with the use of different cathode catalysts (Pd/C or Ir/C).

Graphical Abstract
Introduction
Catalytic hydrogenation of α,β-enones is a significant transformation in organic synthesis [1]. Hydrogenation of enones can give ketones, allyl alcohols, and saturated alcohols, and the control of the chemoselectivity is important. Therefore, there have been numerous studies on the hydrogenation of enones using homogeneous and heterogeneous catalysts.
Meanwhile, electrochemical systems using a proton-exchange membrane (PEM) reactor have been shown to be powerful tools for electrochemical hydrogenation [2-21]. A PEM reactor consists of a membrane called a membrane electrode assembly (MEA), which can act as supporting electrolyte, electrode, and heterogeneous catalyst. Therefore, the further addition of a supporting electrolyte is not necessary for the electrochemical reactions using a PEM reactor, which offers clean and environmentally benign organic transformations. Despite these advantages, the utility of PEM reactors in precise organic synthesis has long been unclear. Recently, however, Atobe and co-workers showed that PEM reactors can be used as a powerful and novel tool for precise organic synthesis [22-26]. For instance, they recently reported a stereoselective reduction of alkynes to Z-alkenes using a PEM reactor. The use of a Pd/C cathode catalyst and the appropriate cathode potential realize the selective synthesis of Z-alkenes [22-24]. They also reported the stereoselective hydrogenation of α,β-unsaturated acids [25] and the reduction of benzoic acids [26].
We have been interested in electrochemical transformations for a long time [27-31] and are paying the most attention to the utility of PEM reactors for organic syntheses, especially chemoselective transformations. In our research, we examined the hydrogenation of enones using a PEM reactor. The designed process is illustrated in Scheme 1. Humidified hydrogen gas is passed through the anodic chamber and substrate is passed through the cathodic chamber. The hydrogen molecules are anodically oxidized to two protons. Then, they move to the cathodic chamber and are reduced by the catalyst of the MEA to monoatomic hydrogen species (adsorbed hydrogen, Had) [22]. Thus generated Had reduces enones 1 to give the corresponding hydrogenated products (ketones 2 and alcohols 3). The expected advantage of PEM reactors is that the reactivity of Had should be controllable by the cathode catalyst and electrochemical parameters. Fortunately, we found that chemoselective reduction of enones 1 can be carried out using different cathode catalysts (Pd/C or Ir/C).
![[1860-5397-18-107-i1]](/bjoc/content/inline/1860-5397-18-107-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Designed electrochemical hydrogenation of enones 1 with a PEM reactor.
Scheme 1: Designed electrochemical hydrogenation of enones 1 with a PEM reactor.
Results and Discussion
Electroreduction of enones to ketones
First, we chose cyclohex-2-en-1-one (1a) as a model compound, and the electroreduction of 1a was carried out using a PEM reactor (Figure 1a, a single path). Pd/C was used as a cathode catalyst. Without electricity, trace amounts of cyclohexanone (2a) and cyclohexanol (3a) were obtained (Table 1, entry 1). With a current of 2.5 mA⋅cm−1, 2a and 3a were obtained in a yield of 3% (current efficiency 66%) and 0.57% (current efficiency 5.7%), respectively (Table 1, entry 2). While 2a was obtained with moderate current efficiency, the yield was far from satisfactory. Therefore, electroreduction with a higher current density was examined (Table 1, entries 3–7). The yield of 2a increased with an increase in the current density (22% yield, 50 mA⋅cm−1).
![[1860-5397-18-107-1]](/bjoc/content/figures/1860-5397-18-107-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Electrochemical setup of the PEM reactor: a) Electrochemical reduction system with the PEM reactor. b) Circulating electrochemical reduction system with the PEM reactor.
Figure 1: Electrochemical setup of the PEM reactor: a) Electrochemical reduction system with the PEM reactor....
Table 1: Effect of the current density on the electrochemical hydrogenation of 1a with a PEM reactor (a single path).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-107-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | current density (mA⋅cm−2) | conversion (%) | yield (efficiency, %)b | selectivity of 2a (%) | |
| 2a | 3a | ||||
| 1 | 0 | 8 | 2 | 0.43 | 84 |
| 2 | 2.5 | <5 | 3 (66) | 0.57 (5.7) | 85 |
| 3 | 5 | <5 | 4 (43) | 0.52 (2.6) | 89 |
| 4 | 10 | 10 | 6 (29) | 0.52 (1.3) | 92 |
| 5 | 12.5 | 15 | 7 (27) | 0.50 (1.0) | 93 |
| 6 | 25 | 19 | 12 (23) | 0.46 (0.5) | 96 |
| 7 | 50 | 28 | 22 (23) | 0.50 (0.3) | 98 |
aReaction conditions: anode catalyst Pt/C, cathode catalyst Pd/C, concentration of 1a 1.0 M, solvent dichloromethane, flow rate of the solution of 1a 0.25 mL⋅min−1, flow rate of H2 gas 500 mL⋅min−1, reaction temperature room temperature. bDetermined by GC analysis using n-dodecane as an internal standard. Values in parentheses are the current efficiency.
To improve the conversion, we designed a circulating system for the PEM reactor (Figure 1b) and used it for the electroreduction of 1a (Table 2). First, we carried out the electroreduction of 1a with a current of 12.5 mA⋅cm−1. As expected, 1a was almost entirely consumed after the passage of 2.0 F⋅mol−1, and 2a was obtained in 67% yield as a major product (Table 2, entry 1). The yield of 2a and 3a was almost the same with a current of 25 mA⋅cm−1 (Table 2, entry 2). Further, the conversion of 1a decreased to 82% with a current of 50 mA⋅cm−1, but 2a was obtained in 64% yield with a similar current efficiency (64%). When the reaction was performed in cyclopentyl methyl ether (CPME) as a solvent, the yield of 2a decreased to 54%, and 3a was obtained in 11% yield (Table 2, entry 4).
Table 2: Effect of the current density and solvent on the electrochemical hydrogenation of 1a with a circulating PEM reactor.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-107-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | current density (mA⋅cm−2) | conversion (%) | yield (efficiency, %)b |
selectivity of 2a
(%) |
|
| 2a | 3a | ||||
| 1 | 12.5 | 99 | 67 (67) | 5 (11) | 93 |
| 2 | 25 | 96 | 63 (63) | 3 (5) | 95 |
| 3 | 50 | 82 | 64 (64) | 2 (4) | 97 |
| 4c | 50 | 82 | 54 (54) | 11 (22) | 83 |
aReaction conditions: anode catalyst Pt/C, concentration of 1a 1.0 M, solvent dichloromethane, flow rate of the solution of 1a 0.25 mL⋅min−1, flow rate of H2 gas,100 mL⋅min−1, reaction temperature room temperature, current density 50 mA⋅cm−2. The solution was circulated until the passage of 2.0 F⋅mol−1. bDetermined by GC analysis using n-dodecane as an internal standard. Values in parentheses show the current efficiency. cPerformed in CPME instead of dichloromethane.
Next, the effect of cathode catalysts was investigated (Table 3). With Ru catalyst, further reduction of the carbonyl group proceeded, and both 2a (32% yield) and 3a (14% yield) were obtained (Table 3, entry 2). With Rh catalyst, the conversion was up to 81%, while the yield of 2a was similar to that with Ru catalyst (Table 3, entry 3). Similarly, both 2a and 3a were obtained with Ir and Pt catalyst (Table 3, entries 4 and 5). In particular, 3a was obtained preferentially with the Ir catalyst. These results revealed that the cathode catalysts strongly affected the selectivity between 2a and 3a. Pd was the best catalyst for the selective synthesis of 2a, and Ir catalyst should be suitable for the formation of 3a, regarding the current efficiency and selectivity (Table 3, entry 4).
Table 3: Effect of catalysts on the electrochemical hydrogenation of 1a with a circulating PEM reactor.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-107-i8.svg?max-width=637&scale=1.0)
|
|||||
| entry | cathode catalyst | conversion (%) | yield (efficiency, %)b | selectivity of 2a (%) | |
| 2a | 3a | ||||
| 1 | Pd/C | 82 | 64 (64) | 2 (4) | 97 |
| 2 | Ru/C | 59 | 32 (32) | 14 (28) | 70 |
| 3 | Rh/C | 81 | 37 (37) | 19 (39) | 66 |
| 4 | Ir/C | 65 | 24 (24) | 26 (52) | 48 |
| 5 | Pt/C | 52 | 11 (12) | 27 (56) | 29 |
aReaction conditions: anode catalyst Pt/C, concentration of 1a 1.0 M, solvent dichloromethane, flow rate of the solution of 1a 0.25 mL⋅min−1, flow rate of H2 gas 100 mL⋅min−1, reaction temperature room temperature, current density 50 mA⋅cm−2. The solution was circulated until the passage of 2.0 F⋅mol−1. bDetermined by GC analysis using n-dodecane as an internal standard. Values in parentheses show the current efficiency.
We also observed reaction profiles of the hydrogenation of 1a with the use of a Pd/C and Ir/C cathode catalyst, respectively (Figure 2). When a Pd/C catalyst was used, 1a was hydrogenated to 2a selectively, and further reduction to 3a was almost completely suppressed (Figure 2a). In contrast, the use of an Ir/C catalyst afforded both 2a and 3a, and generated 2a was smoothly reduced to 3a by further electrolysis (Figure 2b).
![[1860-5397-18-107-2]](/bjoc/content/figures/1860-5397-18-107-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reaction profile of the electrochemical hydrogenation of 1a with a PEM reactor using a) Pd/C and b) Ir/C cathode catalyst. The yield of 2a and 3a and the recovery of 1a are shown in red, brown, and blue, respectively.
Figure 2: Reaction profile of the electrochemical hydrogenation of 1a with a PEM reactor using a) Pd/C and b)...
As mentioned above, ketone 2a was obtained selectively with the use of a Pd/C catalyst for the cathode (Table 3, entry 1). To clarify the scope of the reaction, we carried out the electrochemical reduction of several enones 1 using Pd/C cathode catalyst (Scheme 2). After current was passed to the circulating system until 1a was consumed, the ketone 2a, obtained by the exclusive reduction of the C=C moiety, was obtained in 81% yield with a chemoselectivity of 92%. Similarly, cyclopentanone 2b was obtained from the corresponding enone 1b in 74% yield (88% selectivity). Substituted cyclohexanone such as 3-methylcyclohex-2-en-1-one (1c) gave the desired product 2c selectively in 89% yield (100% selectivity). A benzene-conjugated ketone 1d and an ester 1e could also be subjected to electroreduction to afford the corresponding ketones 2d and 2e in 63% and quantitative yield, respectively. Linear enone 1f gave the desired ketone 2f in high yield (88%, 97% selectivity). Reduction with the PEM reactor also proceeded smoothly with enone 1g, which has a cyclohexene moiety, to give the corresponding ketone 2g in 87% yield (91% selectivity). As shown so far, several kinds of enones 1 could be subjected to electroreduction using the PEM reactor to afford ketones in high yield and selectivity.
![[1860-5397-18-107-i2]](/bjoc/content/inline/1860-5397-18-107-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical hydrogenation of several enones 1 with a circulating PEM reactor using a Pd/C cathode catalyst. Reaction conditions: anode catalyst Pt/C, cathode catalyst Pd/C, concentration of 1 1.0 M, solvent dichloromethane, flow rate of the solution of 1 0.25 mL⋅min−1, flow rate of H2 gas 100 mL⋅min−1, reaction temperature room temperature, current density 50 mA⋅cm−2. Charge was passed to the circulated solution until 1 was consumed. The yield was determined by GC analysis using n-dodecane as an internal standard. Values in parentheses show the chemoselectivity of 3, which was calculated as yield of 2 / yield of (2 + 3).
Scheme 2: Electrochemical hydrogenation of several enones 1 with a circulating PEM reactor using a Pd/C catho...
Electroreduction of enones to saturated alcohols
We next examined the electrochemical reduction of several enones 1 to saturated alcohols 3 using an Ir/C catalyst for the cathode (Scheme 3). Full conversion of 1a under the indicated conditions gave 3a in 79% yield with 98% selectivity. In contrast, electroreduction of cyclopent-2-en-1-one (1b) gave cyclopentanol 3b in 29% yield (46% selectivity), but the reason has not been elaborated yet. With 3-methyl-2-cyclohexen-1-one (1c), alcohol 3c was obtained 72% yield with good selectivity (89%). Both 1f and 1g could be used in this reactions, and the corresponding alcohols 3f and 3g were obtained as major products.
![[1860-5397-18-107-i3]](/bjoc/content/inline/1860-5397-18-107-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Electrochemical hydrogenation of several enones 1 with a circulating PEM reactor using an Ir/C cathode catalyst. Reaction conditions: anode catalyst Pt/C, cathode catalyst Ir/C, concentration of 1 1.0 M, solvent dichloromethane, flow rate of the solution of 1 0.25 mL⋅min−1, flow rate of H2 gas 100 mL⋅min−1, reaction temperature room temperature, current density 50 mA⋅cm−2. Charge was passed to the circulated solution until 1 was consumed. The yield was determined by GC analysis using n-dodecane as an internal standard. Values in parentheses show the chemoselectivity of 3, which was calculated as yield of 3 / yield of (2 + 3).
Scheme 3: Electrochemical hydrogenation of several enones 1 with a circulating PEM reactor using an Ir/C cath...
Mechanistic studies
To gain further insight into the reaction mechanism of the chemoselectivity of a Pd/C cathode system, some additional reactions were carried out (Scheme 4). Electroreduction of 4a as a starting material was carried out using the circulating PEM reactor equipped with a Pd/C cathode. Compound 4a has not been observed under the standard reaction conditions performed so far. The reduction of 4a did not proceed efficiently. Compound 3a was obtained as a major product (26% yield) and 2a was obtained in 6% yield (Scheme 4a). Hydrogenation of the alkene moiety of 4a would proceed selectively, and 2a would be generated via a transfer hydrogenation reaction from 3a as a hydrogen donor [32-35]. These results suggest that electroreduction of 1a would afford 2a directly and not via 4a. Electroreduction of acetophenone did not proceed efficiently, and 42.5% (GC ratio) of acetophenone was recovered with ethylbenzene as a major reduced product (Scheme 4b). We assumed that the reduction would proceed via an enol or enolate intermediate. The reduction of benzophenone also did not proceed smoothly, and only 13% of benzophenone was converted. These results suggest that a Pd/C cathode significantly targets an alkene moiety over a carbonyl group, predominantly leading to the reduction of the C=C moiety.
![[1860-5397-18-107-i4]](/bjoc/content/inline/1860-5397-18-107-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Finally, the electroreduction of 1a was carried out with the use of H2O as a proton source by the PEM reactor with a Pd/C cathode catalyst (Scheme 5). Similar to the reaction with H2, the electroreduction proceeded with high chemoselectivity, and the desired ketone 2a was obtained in 70% yield, whereas alcohol 3a was not observed. Interestingly, the generation of phenol was observed (7% yield), probably because 1a could serve as a hydrogen donor due to the low concentration of hydrogen [32].
![[1860-5397-18-107-i5]](/bjoc/content/inline/1860-5397-18-107-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Electroreduction of 1a with the circulating PEM reactor using H2O as a proton source.
Scheme 5: Electroreduction of 1a with the circulating PEM reactor using H2O as a proton source.
Conclusion
In conclusion, we have developed a system for the electroreduction of enones using a PEM reactor. The reactions proceeded under mild conditions, and highly chemoselective reductions were achieved with the use of appropriate cathode catalysts.
The use of a Pd/C cathode gave carbonyl compounds selectively. In contrast, saturated alcohols were obtained selectively with an Ir/C cathode. The reaction with H2O as a proton source was also achieved. With this reaction system, chemoselective reduction can be performed using only electricity and water, and the product can be easily obtained by simply concentrating the solution coming out of the outlet of the flow system. We are currently trying to reduce various functional groups using this system and shall report the results at a later time.
Supporting Information
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 571.4 KB | Download |
Acknowledgements
The authors thank Prof. Mahito Atobe (Yokohama National University) and his group members for fruitful discussions and suggestions. The authors also thank Prof. Yuta Nishina (Okayama University) and his group members for the help with making the MEA. We also thank Mr. Atsushi Ohsaki in our group for his help.
References
-
Baba, A.; Yasuda, M.; Nishimoto, Y. Partial Reduction of Enones, Styrenes, and Related Systems. In Partial reduction of enones, styrenes, and related systems; Molander, G. A.; Knochel, P., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 8, pp 673–740. doi:10.1016/b978-0-08-097742-3.00822-3
Return to citation in text: [1] -
Hicks, M. T.; Fedkiw, P. S. J. Electrochem. Soc. 1998, 145, 3728–3734. doi:10.1149/1.1838866
Return to citation in text: [1] -
Pintauro, P. N.; Gil, M. P.; Warner, K.; List, G.; Neff, W. Ind. Eng. Chem. Res. 2005, 44, 6188–6195. doi:10.1021/ie0490738
Return to citation in text: [1] -
Sedighi, S.; Gardner, C. L. Electrochim. Acta 2010, 55, 1701–1708. doi:10.1016/j.electacta.2009.10.053
Return to citation in text: [1] -
Zhang, R.; Weidner, J. W. ECS Trans. 2010, 28 (26), 51–63. doi:10.1149/1.3501095
Return to citation in text: [1] -
Fonocho, R.; Gardner, C. L.; Ternan, M. Electrochim. Acta 2012, 75, 171–178. doi:10.1016/j.electacta.2012.04.116
Return to citation in text: [1] -
Green, S. K.; Tompsett, G. A.; Kim, H. J.; Kim, W. B.; Huber, G. W. ChemSusChem 2012, 5, 2410–2420. doi:10.1002/cssc.201200416
Return to citation in text: [1] -
Ehteshami, S. M. M.; Zhou, W. J.; Chan, S. H. Int. J. Hydrogen Energy 2013, 38, 188–196. doi:10.1016/j.ijhydene.2012.10.043
Return to citation in text: [1] -
Stuve, E. M.; Spies, K. A. ECS Trans. 2013, 58 (1), 1723–1731. doi:10.1149/05801.1723ecst
Return to citation in text: [1] -
Stuve, E. M.; Spies, K. ECS Trans. 2013, 53 (9), 21–28. doi:10.1149/05309.0021ecst
Return to citation in text: [1] -
Ehteshami, S. M. M.; Vignesh, S.; Rasheed, R. K. A.; Chan, S. H. Appl. Energy 2016, 170, 388–393. doi:10.1016/j.apenergy.2016.03.001
Return to citation in text: [1] -
Ogihara, H.; Maezuru, T.; Ogishima, Y.; Yamanaka, I. ChemistrySelect 2016, 1, 5533–5537. doi:10.1002/slct.201601082
Return to citation in text: [1] -
Ogihara, H.; Maezuru, T.; Ogishima, Y.; Yamanaka, I. Electrocatalysis 2018, 9, 220–225. doi:10.1007/s12678-017-0419-1
Return to citation in text: [1] -
Caravaca, A.; Garcia-Lorefice, W. E.; Gil, S.; de Lucas-Consuegra, A.; Vernoux, P. Electrochem. Commun. 2019, 100, 43–47. doi:10.1016/j.elecom.2019.01.016
Return to citation in text: [1] -
Carl, S.; Waldrop, K.; Pintauro, P.; Thompson, L. T.; Tarpeh, W. A. ChemElectroChem 2019, 6, 5563–5570. doi:10.1002/celc.201901314
Return to citation in text: [1] -
Sato, M.; Ogihara, H.; Yamanaka, I. ISIJ Int. 2019, 59, 623–627. doi:10.2355/isijinternational.isijint-2018-551
Return to citation in text: [1] -
Takano, K.; Tateno, H.; Matsumura, Y.; Fukazawa, A.; Kashiwagi, T.; Nakabayashi, K.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Chem. Lett. 2016, 45, 1437–1439. doi:10.1246/cl.160766
Return to citation in text: [1] -
Takano, K.; Tateno, H.; Matsumura, Y.; Fukazawa, A.; Kashiwagi, T.; Nakabayashi, K.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Bull. Chem. Soc. Jpn. 2016, 89, 1178–1183. doi:10.1246/bcsj.20160165
Return to citation in text: [1] -
Fukazawa, A.; Takano, K.; Matsumura, Y.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Bull. Chem. Soc. Jpn. 2018, 91, 897–899. doi:10.1246/bcsj.20180021
Return to citation in text: [1] -
Egbert, J. D.; Thomsen, E. C.; O’Neill-Slawecki, S. A.; Mans, D. M.; Leitch, D. C.; Edwards, L. J.; Wade, C. E.; Weber, R. S. Org. Process Res. Dev. 2019, 23, 1803–1812. doi:10.1021/acs.oprd.8b00379
Return to citation in text: [1] -
Liu, J.; Chen, R.; Zhu, X.; Liao, Q.; Ye, D.; Zhang, B.; Liu, M.; Chen, G.; Wang, K. RSC Adv. 2019, 9, 23560–23569. doi:10.1039/c9ra02648g
Return to citation in text: [1] -
Fukazawa, A.; Minoshima, J.; Tanaka, K.; Hashimoto, Y.; Kobori, Y.; Sato, Y.; Atobe, M. ACS Sustainable Chem. Eng. 2019, 7, 11050–11055. doi:10.1021/acssuschemeng.9b01882
Return to citation in text: [1] [2] [3] -
Nogami, S.; Nagasawa, K.; Fukazawa, A.; Tanaka, K.; Mitsushima, S.; Atobe, M. J. Electrochem. Soc. 2020, 167, 155506. doi:10.1149/1945-7111/abaae7
Return to citation in text: [1] [2] -
Nogami, S.; Shida, N.; Iguchi, S.; Nagasawa, K.; Inoue, H.; Yamanaka, I.; Mitsushima, S.; Atobe, M. ACS Catal. 2022, 12, 5430–5440. doi:10.1021/acscatal.2c01594
Return to citation in text: [1] [2] -
Fukazawa, A.; Tanaka, K.; Hashimoto, Y.; Sato, Y.; Atobe, M. Electrochem. Commun. 2020, 115, 106734. doi:10.1016/j.elecom.2020.106734
Return to citation in text: [1] [2] -
Fukazawa, A.; Shimizu, Y.; Shida, N.; Atobe, M. Org. Biomol. Chem. 2021, 19, 7363–7368. doi:10.1039/d1ob01197a
Return to citation in text: [1] [2] -
Yoshida, J.-i.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112
Return to citation in text: [1] -
Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J.-i. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p
Return to citation in text: [1] -
Mitsudo, K.; Yamamoto, J.; Akagi, T.; Yamashita, A.; Haisa, M.; Yoshioka, K.; Mandai, H.; Ueoka, K.; Hempel, C.; Yoshida, J.-i.; Suga, S. Beilstein J. Org. Chem. 2018, 14, 1192–1202. doi:10.3762/bjoc.14.100
Return to citation in text: [1] -
Mitsudo, K.; Matsuo, R.; Yonezawa, T.; Inoue, H.; Mandai, H.; Suga, S. Angew. Chem., Int. Ed. 2020, 59, 7803–7807. doi:10.1002/anie.202001149
Return to citation in text: [1] -
Kurimoto, Y.; Yamashita, J.; Mitsudo, K.; Sato, E.; Suga, S. Org. Lett. 2021, 23, 3120–3124. doi:10.1021/acs.orglett.1c00807
Return to citation in text: [1] -
Brieger, G.; Nestrick, T. J. Chem. Rev. 1974, 74, 567–580. doi:10.1021/cr60291a003
Return to citation in text: [1] [2] -
Bagnell, L.; Strauss, C. R. Chem. Commun. 1999, 287–288. doi:10.1039/a808977i
Return to citation in text: [1] -
Albrecht, M.; Crabtree, R. H.; Mata, J.; Peris, E. Chem. Commun. 2002, 32–33. doi:10.1039/b109491b
Return to citation in text: [1] -
Hillier, A. C.; Lee, H. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2001, 20, 4246–4252. doi:10.1021/om0103456
Return to citation in text: [1]
| 1. | Baba, A.; Yasuda, M.; Nishimoto, Y. Partial Reduction of Enones, Styrenes, and Related Systems. In Partial reduction of enones, styrenes, and related systems; Molander, G. A.; Knochel, P., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 8, pp 673–740. doi:10.1016/b978-0-08-097742-3.00822-3 |
| 25. | Fukazawa, A.; Tanaka, K.; Hashimoto, Y.; Sato, Y.; Atobe, M. Electrochem. Commun. 2020, 115, 106734. doi:10.1016/j.elecom.2020.106734 |
| 22. | Fukazawa, A.; Minoshima, J.; Tanaka, K.; Hashimoto, Y.; Kobori, Y.; Sato, Y.; Atobe, M. ACS Sustainable Chem. Eng. 2019, 7, 11050–11055. doi:10.1021/acssuschemeng.9b01882 |
| 23. | Nogami, S.; Nagasawa, K.; Fukazawa, A.; Tanaka, K.; Mitsushima, S.; Atobe, M. J. Electrochem. Soc. 2020, 167, 155506. doi:10.1149/1945-7111/abaae7 |
| 24. | Nogami, S.; Shida, N.; Iguchi, S.; Nagasawa, K.; Inoue, H.; Yamanaka, I.; Mitsushima, S.; Atobe, M. ACS Catal. 2022, 12, 5430–5440. doi:10.1021/acscatal.2c01594 |
| 22. | Fukazawa, A.; Minoshima, J.; Tanaka, K.; Hashimoto, Y.; Kobori, Y.; Sato, Y.; Atobe, M. ACS Sustainable Chem. Eng. 2019, 7, 11050–11055. doi:10.1021/acssuschemeng.9b01882 |
| 23. | Nogami, S.; Nagasawa, K.; Fukazawa, A.; Tanaka, K.; Mitsushima, S.; Atobe, M. J. Electrochem. Soc. 2020, 167, 155506. doi:10.1149/1945-7111/abaae7 |
| 24. | Nogami, S.; Shida, N.; Iguchi, S.; Nagasawa, K.; Inoue, H.; Yamanaka, I.; Mitsushima, S.; Atobe, M. ACS Catal. 2022, 12, 5430–5440. doi:10.1021/acscatal.2c01594 |
| 25. | Fukazawa, A.; Tanaka, K.; Hashimoto, Y.; Sato, Y.; Atobe, M. Electrochem. Commun. 2020, 115, 106734. doi:10.1016/j.elecom.2020.106734 |
| 26. | Fukazawa, A.; Shimizu, Y.; Shida, N.; Atobe, M. Org. Biomol. Chem. 2021, 19, 7363–7368. doi:10.1039/d1ob01197a |
| 2. | Hicks, M. T.; Fedkiw, P. S. J. Electrochem. Soc. 1998, 145, 3728–3734. doi:10.1149/1.1838866 |
| 3. | Pintauro, P. N.; Gil, M. P.; Warner, K.; List, G.; Neff, W. Ind. Eng. Chem. Res. 2005, 44, 6188–6195. doi:10.1021/ie0490738 |
| 4. | Sedighi, S.; Gardner, C. L. Electrochim. Acta 2010, 55, 1701–1708. doi:10.1016/j.electacta.2009.10.053 |
| 5. | Zhang, R.; Weidner, J. W. ECS Trans. 2010, 28 (26), 51–63. doi:10.1149/1.3501095 |
| 6. | Fonocho, R.; Gardner, C. L.; Ternan, M. Electrochim. Acta 2012, 75, 171–178. doi:10.1016/j.electacta.2012.04.116 |
| 7. | Green, S. K.; Tompsett, G. A.; Kim, H. J.; Kim, W. B.; Huber, G. W. ChemSusChem 2012, 5, 2410–2420. doi:10.1002/cssc.201200416 |
| 8. | Ehteshami, S. M. M.; Zhou, W. J.; Chan, S. H. Int. J. Hydrogen Energy 2013, 38, 188–196. doi:10.1016/j.ijhydene.2012.10.043 |
| 9. | Stuve, E. M.; Spies, K. A. ECS Trans. 2013, 58 (1), 1723–1731. doi:10.1149/05801.1723ecst |
| 10. | Stuve, E. M.; Spies, K. ECS Trans. 2013, 53 (9), 21–28. doi:10.1149/05309.0021ecst |
| 11. | Ehteshami, S. M. M.; Vignesh, S.; Rasheed, R. K. A.; Chan, S. H. Appl. Energy 2016, 170, 388–393. doi:10.1016/j.apenergy.2016.03.001 |
| 12. | Ogihara, H.; Maezuru, T.; Ogishima, Y.; Yamanaka, I. ChemistrySelect 2016, 1, 5533–5537. doi:10.1002/slct.201601082 |
| 13. | Ogihara, H.; Maezuru, T.; Ogishima, Y.; Yamanaka, I. Electrocatalysis 2018, 9, 220–225. doi:10.1007/s12678-017-0419-1 |
| 14. | Caravaca, A.; Garcia-Lorefice, W. E.; Gil, S.; de Lucas-Consuegra, A.; Vernoux, P. Electrochem. Commun. 2019, 100, 43–47. doi:10.1016/j.elecom.2019.01.016 |
| 15. | Carl, S.; Waldrop, K.; Pintauro, P.; Thompson, L. T.; Tarpeh, W. A. ChemElectroChem 2019, 6, 5563–5570. doi:10.1002/celc.201901314 |
| 16. | Sato, M.; Ogihara, H.; Yamanaka, I. ISIJ Int. 2019, 59, 623–627. doi:10.2355/isijinternational.isijint-2018-551 |
| 17. | Takano, K.; Tateno, H.; Matsumura, Y.; Fukazawa, A.; Kashiwagi, T.; Nakabayashi, K.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Chem. Lett. 2016, 45, 1437–1439. doi:10.1246/cl.160766 |
| 18. | Takano, K.; Tateno, H.; Matsumura, Y.; Fukazawa, A.; Kashiwagi, T.; Nakabayashi, K.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Bull. Chem. Soc. Jpn. 2016, 89, 1178–1183. doi:10.1246/bcsj.20160165 |
| 19. | Fukazawa, A.; Takano, K.; Matsumura, Y.; Nagasawa, K.; Mitsushima, S.; Atobe, M. Bull. Chem. Soc. Jpn. 2018, 91, 897–899. doi:10.1246/bcsj.20180021 |
| 20. | Egbert, J. D.; Thomsen, E. C.; O’Neill-Slawecki, S. A.; Mans, D. M.; Leitch, D. C.; Edwards, L. J.; Wade, C. E.; Weber, R. S. Org. Process Res. Dev. 2019, 23, 1803–1812. doi:10.1021/acs.oprd.8b00379 |
| 21. | Liu, J.; Chen, R.; Zhu, X.; Liao, Q.; Ye, D.; Zhang, B.; Liu, M.; Chen, G.; Wang, K. RSC Adv. 2019, 9, 23560–23569. doi:10.1039/c9ra02648g |
| 32. | Brieger, G.; Nestrick, T. J. Chem. Rev. 1974, 74, 567–580. doi:10.1021/cr60291a003 |
| 33. | Bagnell, L.; Strauss, C. R. Chem. Commun. 1999, 287–288. doi:10.1039/a808977i |
| 34. | Albrecht, M.; Crabtree, R. H.; Mata, J.; Peris, E. Chem. Commun. 2002, 32–33. doi:10.1039/b109491b |
| 35. | Hillier, A. C.; Lee, H. M.; Stevens, E. D.; Nolan, S. P. Organometallics 2001, 20, 4246–4252. doi:10.1021/om0103456 |
| 22. | Fukazawa, A.; Minoshima, J.; Tanaka, K.; Hashimoto, Y.; Kobori, Y.; Sato, Y.; Atobe, M. ACS Sustainable Chem. Eng. 2019, 7, 11050–11055. doi:10.1021/acssuschemeng.9b01882 |
| 27. | Yoshida, J.-i.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112 |
| 28. | Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J.-i. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p |
| 29. | Mitsudo, K.; Yamamoto, J.; Akagi, T.; Yamashita, A.; Haisa, M.; Yoshioka, K.; Mandai, H.; Ueoka, K.; Hempel, C.; Yoshida, J.-i.; Suga, S. Beilstein J. Org. Chem. 2018, 14, 1192–1202. doi:10.3762/bjoc.14.100 |
| 30. | Mitsudo, K.; Matsuo, R.; Yonezawa, T.; Inoue, H.; Mandai, H.; Suga, S. Angew. Chem., Int. Ed. 2020, 59, 7803–7807. doi:10.1002/anie.202001149 |
| 31. | Kurimoto, Y.; Yamashita, J.; Mitsudo, K.; Sato, E.; Suga, S. Org. Lett. 2021, 23, 3120–3124. doi:10.1021/acs.orglett.1c00807 |
| 26. | Fukazawa, A.; Shimizu, Y.; Shida, N.; Atobe, M. Org. Biomol. Chem. 2021, 19, 7363–7368. doi:10.1039/d1ob01197a |
| 32. | Brieger, G.; Nestrick, T. J. Chem. Rev. 1974, 74, 567–580. doi:10.1021/cr60291a003 |
© 2022 Mitsudo et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.