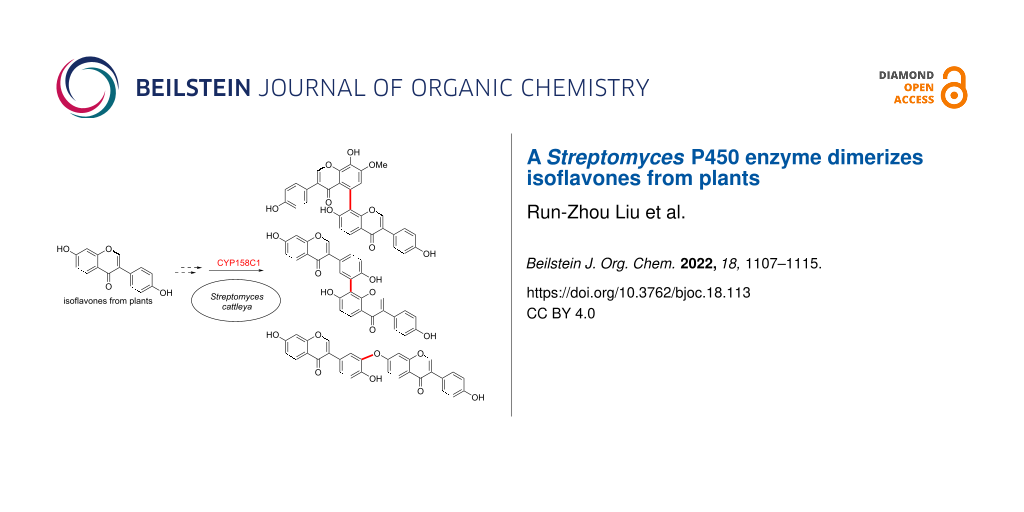
Abstract
Dimerization is a widespread natural strategy that enables rapid structural diversification of natural products. However, our understanding of the dimerization enzymes involved in this biotransformation is still limited compared to the numerous reported dimeric natural products. Here, we report the characterization of three new isoflavone dimers from Streptomyces cattleya cultured on an isoflavone-containing agar plate. We further identified a cytochrome P450 monooxygenase, CYP158C1, which is able to catalyze the dimerization of isoflavones. CYP158C1 can also dimerize plant-derived polyketides, such as flavonoids and stilbenes. Our work represents a unique bacterial P450 that can dimerize plant polyphenols, which extends the insights into P450-mediated biaryl coupling reactions in biosynthesis.

Graphical Abstract
Introduction
Dimerization is a ubiquitous biotransformation in nature. Almost all forms of life, including bacteria, fungi, and plants, have the ability to produce homo- or heterodimeric natural products, which enables rapid structural diversification from simple monomers [1]. Often, the dimerized products exhibit significant biological activities due to the increased functional group density or complex stereochemistry, as seen in vinblastine [2], julichrome [3], himastatin [4], and biflavone [5]. However, the structural complexity of the dimeric natural products has hampered synthetic chemistry approaches towards these molecules, since chemo-, regio-, and atroposelective formation of the biaryl linkage remains highly challenging and often requires prefunctionalization of the substrate monomers, costly metal ligands, or tedious protection–deprotection steps [6-9].
With the advance of biosynthetic studies on natural products, a number of enzyme classes that are responsible for the dimer formation have been identified [1,10-14]. In plants and fungi, laccases and cytochrome P450 monooxygenases play pivotal roles in the biaryl bond formation of various polyketide dimers [10,15,16]. In contrast, in bacteria, P450 enzymes are the dominant catalysts, but no laccases have been reported for dimerization reactions (Figure S1, Supporting Information File 1). Due to the high reaction selectivity that the enzyme active site offers, these enzymes provide biocatalytic means for the biaryl linkage formation, and recent enzyme engineering efforts also demonstrated selective and efficient production of unnatural dimers or cross-coupling products, starting from simple monomers [17-19]. Nevertheless, our knowledge of enzyme-mediated dimerization is still limited in contrast to the numerous reported dimeric natural products.
Phenol coupling in plant polyphenol biosynthesis is one of the earliest documented biocatalytic dimerization reactions [20]. Contrary to flavone dimers and oligomers being abundant in nature, only limited dimeric compounds have been reported for isoflavones. Isoflavones bear a characteristic 3-phenylchroman skeleton, which is formed by the B-ring migration from the flavonoid scaffold catalyzed by isoflavone synthases [21]. The sporadic distribution of this isoflavone synthase limits the discovery of isoflavones in the plant kingdom [22], and the enzymes catalyzing isoflavone dimerization, to our knowledge, remain uncharacterized.
In this study, we report the discovery of a Streptomyces cytochrome P450 enzyme that catalyzes dimerization of plant isoflavones. By untargeted metabolomics, we isolated three new isoflavone dimers, namely cattleyaisoflavones A–C (1–3), from Streptomyces cattleya cultured in soy flour-containing media. We then identified a P450 enzyme, CYP158C1, which is able to dimerize the isoflavones as well as other plant-derived phenolic compounds. This work extends the insight on the substrate scope of P450-mediated dimerization and provides a potential biocatalytic tool for the synthesis of isoflavone dimers in future.
Results and Discussion
Characterization of three isoflavone dimers derived from daidzein (4)
During our effort of untargeted metabolomic screening of Streptomyces species, we identified several unknown isoflavone-like metabolites from S. cattleya NRRL8057 cultured on mannitol soya flour (MS) agar plates (Figure S2, Supporting Information File 1). Purification of the three major unknown compounds resulted in the isolation of the isoflavone dimers cattleyaisoflavones A (1), B (2), and C (3) (Figure 1). HRMS analyses suggested the molecular formula of both 2 and 3 to be C30H18O8 and that of 1 to be C31H20O9 (Figure S3, Supporting Information File 1), which matches the molecular formula of isoflavone dimers or dimeric derivatives.
![[1860-5397-18-113-1]](/bjoc/content/figures/1860-5397-18-113-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of cattleyaisoflavones A (1), B (2), C (3), and daidzein (4).
Figure 1: Structures of cattleyaisoflavones A (1), B (2), C (3), and daidzein (4).
To confirm whether 1–3 were biosynthesized de novo by the strain or derived from the media that contain isoflavones, we cultured S. cattleya on International Streptomyces Project-2 (ISP-2) liquid medium that does not contain isoflavones. These cultures did not produce 1–3, but daidzein (4) supplementation in ISP-2 liquid medium restored the production of 1–3, suggesting that S. cattleya utilizes exogenous isoflavone to form the dimer products 1–3 (Figure 2a).
![[1860-5397-18-113-2]](/bjoc/content/figures/1860-5397-18-113-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) The culture in ISP-2 liquid did not produce 1–3, while feeding with 4 restored the production (analytical method A according to Experimental section, 254 nm). b) Chemical genetics experiments indicated three P450 inhibitors could reduce the production of dimers derived from 4 (analytical method B to better analyze the isoflavone region, 254 nm). CTA: clotrimazole (2.5 µM), FCA: fluconazole (1000 µM), RVT: resveratrol (100 µM).
Figure 2: a) The culture in ISP-2 liquid did not produce 1–3, while feeding with 4 restored the production (a...
A comparison of the 1H NMR spectra of 1 and 4 (Tables S3 and S4, Supporting Information File 1) suggested that 1 has a biaryl linkage between the two A-rings. The 1H,1H-COSY correlation of H-5’’ (δH 8.06, d) and H-6’’ (δH 7.03, d) revealed C-8’’ as one of the coupling sites. The methoxy group (δH 3.98, s) was assigned to the C-7 site because of the 1H,1H-NOESY correlation with H-6 (δH 6.98, s). The HMBC correlations of H-6 to C-8 (δC 136.3), C-10 (δC 118.9), C-7 (δC 151.7), C-5 (δC 123.6), and C-8’’ (δC 119.1) suggested that C-5 is the coupling site on this unit, and C-8 was substituted by a hydroxy group. Finally, the structure of 1 was determined as a dimer with a rare 5,8’’ biaryl coupling skeleton. Likewise, C-8’’ was assigned as one of the coupling sites of 2. Three B-ring protons, H-2’ (δH 7.38, d), H-5’ (δH 7.02, d), and H-6’ (δH 7.48, dd), suggested that C-3’ was another coupling site. Thus, the structure of 2 was established as a 3’,8’’-coupled dimer (Table S5, Supporting Information File 1). For the dimer 3, three hydroxy and fifteen sp2 protons indicated a C–O bond linkage. An analysis of 1H,1H-COSY and HMBC spectra suggested it to be a 3’–O–7’’-coupled dimer (Table S6, Supporting Information File 1).
The absolute configuration of 1 and 2 with respect to atropisomerism was assessed by chiral HPLC analysis and the comparison of experimental and calculated ECD spectra. While 1 showed a ≈3:1 atropisomer ratio, with the M-atropisomer being the major form, 2 did not exhibit a Cotton effect in the ECD measurement, suggesting that it is in a freely rotating state (Supporting Information File 1).
Discovery of the P450 enzyme responsible for dimerization
We next investigated which enzyme in S. cattleya is responsible for dimerization of 4. The genome of S. cattleya (GCF_000237305.1) did not encode any laccase homologs but encoded 41 P450 enzymes in total. To investigate whether P450 enzymes mediate this dimerization reaction, we first employed a chemical genetics screening using three P450 inhibitors, CTA (strong inhibitor), FCA (moderate inhibitor), and RVT (polyphenol inhibitor) [23,24]. All three agents reduced the production of cattleyaisoflavones in a concentration-dependent manner (Figure 2b and Figure S4, Supporting Information File 1), suggesting that P450 enzymes play a key role in isoflavone dimerization.
The known bacterial P450 enzymes involved in biaryl coupling can be classified into four types, including CYP158 enzymes for dimerization of naphthols [25-27], JulI/SetI/SptI clade for anthraquinones [3], HmtS/ClpS/LtzS clade for cyclopeptides [4,18,19], and Bmp7 clade for polybrominated substrates (Table S7 and Figure S1, Supporting Information File 1) [13,17]. These P450 enzymes tend to have broad substrate specificity and form a variety of biaryl products. Thus, we constructed a phylogenetic tree using the 41 P450 enzymes in S. cattleya, together with these reported dimerization P450 enzymes (Figure S5, Supporting Information File 1). Notably, a P450 (WP_014145731.1, here termed CYP158C1) was clustered into the CYP158 clade that reportedly catalyzes dimerization of type III polyketide synthase (T3PKS) products, such as naphthols. Considering the similar biosynthetic pathway of isoflavones shared, this enzyme was expressed in E. coli and purified for in vitro biochemical assay together with four other P450 enzymes (CYP1–4) that were successfully expressed out of 10 P450 enzymes from each representative clade (Figures S5 and S6, Supporting Information File 1). The results showed that only CYP158C1 was able to dimerize 4 (Figure 3 and Figure S7, Supporting Information File 1), indicating that CYP158C1 is the most likely enzyme for isoflavone dimerization. Interestingly, different from the in vivo extract, in vitro experiments gave a major dimer 5, a 3’,3’ coupled dimer assigned by MS–MS (Figures S8 and S14, Supporting Information File 1). Although dimer 3 was not visible in the HPLC analysis, a trace amount of 3 was detected by HRMS analysis (Figure S8, Supporting Information File 1). Redox partners might be responsible for the different regioselectivity between in vivo and in vitro reactions, as it has been reported that different reductases would lead to different diastereomeric dimers of coclaurine [28]. The absence of the product 1 in vitro is reasonable because the biosynthesis of 1 requires an additional methyltransferase from S. cattleya for the methoxy group formation.
![[1860-5397-18-113-3]](/bjoc/content/figures/1860-5397-18-113-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CYP158C1 dimerizes 4 to form dimers 2 and 5 in vitro (analytical method B, 254 nm). Control conditions (green): 4 (0.4 mM), NADPH (1 mM), and ferredoxin–NADP+ reductase SelFdR0978/ferredoxin SelFdx1499 (FdR/Fdx, 5 µM) in buffer (50 mM PBS, 0.2 mM DTT, 1 mM EDTA, pH 7.4) were incubated at 37 °C for 1 h. Red: the conditions above with the addition of CYP158C1 (3 µM). Blue: the conditions above with the addition of CYP158C1 (3 µM) and RVT (0.5 mM).
Figure 3: CYP158C1 dimerizes 4 to form dimers 2 and 5 in vitro (analytical method B, 254 nm). Control conditi...
Substrate scope and reaction mechanism
After identification of CYP158C1 as the isoflavone dimerization enzyme, we next explored the substrate scope of the catalyst using a variety of plant-derived polyketides (Scheme 1). The results revealed that the 5-OH substitution on isoflavone (see compound 6) was tolerable for dimerization (Figure S15, Supporting Information File 1), whereas the additional 4’-methoxyl group at the B-ring (see 7) prevented the dimerization. Two isoflavone glucosides, 8 and 9, failed to be transformed, presumably due to the excessive molecular size. When using the other flavonoids 10–17 as substrates, we observed the corresponding dimeric products for the substrates 10–12 (Figures S16–S18, Supporting Information File 1) but not for 13–17, suggesting that CYP158C1 is also compatible with compounds with flavone scaffold. Apart from flavonoids, this enzyme also dimerized umbelliferone (18), a substrate of the fungal P450 KtnC [14,18], and RVT with relatively low efficiency (Figures S19 and S20, Supporting Information File 1). Other plant polyketides, such as anthraquinones 19 and 20 and phenylpropanoids 21–24, failed to be dimerized.
![[1860-5397-18-113-i1]](/bjoc/content/inline/1860-5397-18-113-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: a) Compatible and b) incompatible substrates of CYP158C1. Products were identified using analytical method D.
Scheme 1: a) Compatible and b) incompatible substrates of CYP158C1. Products were identified using analytical...
The reaction mechanism of P450-mediated phenol dimerization is believed to involve oxidative radical–radical coupling, though other mechanisms, such as radical addition, radical cation addition, and electrophilic aromatic addition, have also been proposed [1,10,29]. A proposed mechanism is depicted in Scheme 2: First, the hydroxy group on the A- or B-ring is converted into a radical by a P450-induced single-electron transformation. The resulting radical then migrates to the π-system and is stabilized in the ortho- and para-positions, generating diverse carbon radical intermediates. As a result, various dimers are formed via promiscuous coupling of these radical intermediates.
![[1860-5397-18-113-i2]](/bjoc/content/inline/1860-5397-18-113-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism of CYP158C1-mediated dimerization of isoflavones.
Scheme 2: Proposed mechanism of CYP158C1-mediated dimerization of isoflavones.
Although we cannot exclude the possibility that other P450 enzymes in S. cattleya may also catalyze isoflavone dimerization, the above results indicated that CYP158C1 dimerizes isoflavones and several plant-derived aromatic polyketides, albeit with low catalytic efficiency. Future optimization of the reductase partner or reaction conditions may be required to improve the enzyme activity. Interestingly, we found that CYP158C1 clusters together with a T3PKS gene in the S. cattleya genome, which is similar to the biflaviolin [30] and naringenin [31] biosynthetic gene clusters. The native function of these type-III polyketide synthase products is believed to be involved in the protection of the host against oxidative radicals generated by UV irradiation [25,32]. To verify the antioxidative effect of the isoflavone dimers, 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS)-based antioxidant activity was performed [33]. The results showed that while 2 and 3 had an activity roughly equal to the monomers 4 and 6, 1 displayed a roughly twofold radical scavenging capacity (Figure S9, Supporting Information File 1). This is in agreement with the previous report that the configuration of the hydroxy group of the B-ring plays a key role in the antioxidant activity [34]. Thus, CYP158C1 might provide an alternative pathway for antioxidant biosynthesis by using exogenous metabolites as substrates. The native substrate for CYP158C1, presumably produced by the clustered T3PKS, has yet to be identified.
The broad substrate specificity of CYP158C1 towards plant polyphenols suggests a potential host–microbial interaction in the rhizosphere. Flavones and isoflavones are common molecules in plant roots and can be secreted in soil, playing a role in allelopathic interference [35,36]. To our knowledge, this is the first bacterial P450 that can dimerize plant flavonoids. Since dimerization of polyphenols in plants is mostly catalyzed by laccases, the dimerization of (iso)flavones by a P450 in Streptomyces may represent a convergent evolution in enzyme chemistry between plants and bacteria.
Conclusion
In conclusion, we have characterized three novel isoflavone dimers from S. cattleya and identified the P450 enzyme CYP158C1 to be able to catalyze biaryl coupling reactions using plant-derived daidzein (4) as a substrate. The enzyme can dimerize various flavonoids and provides a potential synthetic tool for the dimerization of aromatic polyketides.
Experimental
General experimental procedures
NMR spectra were acquired on Bruker 500 and 600 MHz AVANCE NEO spectrometers with TMS as an internal standard. HPLC–UV analysis was performed on an Agilent 1260 Infinity II HPLC system. UHPLC–HRMS analysis was performed on a Waters Synapt-G2-Si UHPLC-ESI-QToF-MS system. UV and CD spectra were acquired under N2 gas by a Chirascan V100 spectrometer (Applied Photophysics). Strain cultivation was performed at 28 °C in an incubator for agar plates or in a rotary shaker at 200 rpm for liquid media.
Strain preparation and fermentation
The strain S. cattleya NRRL 8057 was purchased from the China General Microbiological Culture Collection Center (CGMCC). Seed culture of this strain was done in ISP-2 medium (yeast extract 0.4 g, malt extract 1 g, glucose 0.4 g, distilled water up to 100 mL, pH 7.2) for 2 days. Isoflavone dimer production was performed on MS agar medium (mannitol 20 g, soy flour 20 g, agar 20 g, distilled water up to 1 L) for 15 days from the above seed culture.
Analytical methods
All HPLC analyses were performed using (A) H2O with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid at 40 °C column temperature unless otherwise noted. The HPLC–UV analysis of feeding experiments was conducted with a YMC-Triart C18 ExRS column (150 mm × 3.0 mm, 3 μm, 12 nm, YMC) with a gradient of 5% B (0 min) to 95% B (12 min) at a flow rate of 0.5 mL/min (analytical method A). The HPLC–UV analysis of isoflavones was conducted with a Poroshell 120 SB-C18 column (100 mm × 3.0 mm, 2.7 μm, Agilent) with a gradient of 30% B (0 min) to 45% B (10 min) at a flow rate of 0.5 mL/min (analytical method B). The UHPLC–HRMS analysis of isoflavones was conducted with a HSS T3 column (100 mm × 2.1 mm, 1.8 μm, Waters) with a gradient of 30% B (0 min) to 45% B (4.5 min) at a flow rate of 0.4 mL/min (analytical method C). The UHPLC–HRMS analysis of the substrate scope was conducted with a HSS T3 column (100 mm × 2.1 mm, 1.8 μm, Waters) with a gradient of 0% B (0 min) to 100% B (5.0 min) at a flow rate of 0.4 mL/min (analytical method D). All HRMS analyses were performed in positive model with a scan range of m/z 50–1600. For MS–MS analysis, the collision-induced dissociation (CID) energy was set to 15–40 eV or 30–50 eV depending on the compounds. The HPLC–UV analysis of the chiral separation was conducted with a CHIRALCEL OX-3R column (150 mm × 4.6 mm, 3 μm, DAICEL) with isocratic 35% B at a flow rate of 1.0 mL/min at 30 °C column temperature, and an Agilent G6215B MSD detector was used for peak assignment (analytical method E).
Isolation of compounds
To isolate compounds 1–3, the fermentation culture of S. cattleya on MS agar medium (125 plates, 80 mL/plate each) was collected and extracted using organic solvent (CHCl3/MeOH 1:1, v/v, 20 L in total). After removing the solvent in vacuo, the crude extract was redissolved in methanol and separated by Sephdex LH-20 (GE healthcare) column chromatography using methanol as eluent. The yellow band at the elution tail on the LH-20 column (Figure S2, Supporting Information File 1) was collected and further purified by a Shimadzu LC-20AD semipreparative HPLC by using (A) H2O with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. Compounds 1 (5.0 mg), 2 (2.2 mg), and 3 (2.0 mg) were isolated using a YMC-Triart C18 column (250 mm × 10 mm, 5 μm, YMC) at 4 mL/min with a gradient of 40% B (0 min) to 60% B (12 min). A second round of purification was then performed for 1–3 with isocratic 35%, 38%, and 45% B, respectively, with a YMC-Triart Phenyl column (250 mm × 10 mm, 5 μm, YMC) at 4 mL/min.
Cattleyaisoflavone A (1): yellowish powder; UV (MeOH) λmax (log ε) 257, 304 nm; for NMR spectral data, see Table S4, Supporting Information File 1; HRMS–ESI (m/z): [M + H]+ calcd for C31H20O9, 537.1180; found, 537.1206.
Cattleyaisoflavone B (2): yellowish powder; UV (MeOH) λmax 255, 310 nm; for NMR spectral data, see Table S5, Supporting Information File 1; HRMS–ESI (m/z): [M + H]+ calcd for C30H18O8, 507.1074; found, 507.1091.
Cattleyaisoflavone C (3): white powder; UV (MeOH) λmax 254, 300 nm; for NMR spectral data, see Table S6, Supporting Information File 1; HRMS–ESI (m/z): [M + NH+ calcd for C30H18O8, 507.1074; found, 507.1091.
Feeding experiments
Feeding experiments were performed in 500 mL flasks each containing 100 mL of ISP-2 liquid medium and 10 mg daidzein (4, as 1 mL DMSO solution added after autoclave) by 1% inoculation from the above seed culture. On the 5th day, 2 mL of medium from each sample was taken and freeze-dried. Then, 1 mL MeOH was added and the mixture was treated with ultrasound for 15 min. After centrifugation, the supernatant was directly analyzed by HPLC following analytical method A.
Chemical genetics experiments
All samples involved in inhibition experiments were set up using 100 mL flasks containing 20 mL ISP-2 medium, 0.02 mg/mL daidzein (4), and the P450 inhibitor, by 1% inoculation from the above seed culture. On the 9th day, 0.5 mL of medium from each sample was taken and freeze-dried. Then, 0.2 mL MeOH was added, and the mixture was treated with ultrasound for 15 min. After centrifugation, the supernatant was detected following analytical method B.
Bioinformatic analysis
Multiple sequence alignment was constructed by MAFFT 7.450 using the E-INS-I method [37], and the phylogenetic tree was constructed by FastTree 2.1.11 [38] with default parameters by using Geneious Prime 2022.1.1 (see https://www.geneious.com).
Protein purification and biochemical assays
The Streptomyces genome was extracted by TIANamp Bacteria DNA Kit (TIANGEN BIOTECH), and the P450 genes were amplified by PCR (Table S1, Supporting Information File 1) and cloned into a pET-28a(+) vector by the T5 exonuclease DNA assembly method [39]. The resulting expression vectors were individually introduced into E. coli BL21(DE3) for protein expression. Cells harboring expression plasmid were inoculated into LB medium with 50 µg/mL kanamycin and incubated at 37 °C until the culture reached an OD600 of 0.4–0.6. The cultures were then cooled to 16 °C and induced with 0.1 mM isopropyl β-ᴅ-1-thiogalactopyranoside (IPTG) for 16 h. Cells were harvested by centrifugation (10 min, 8,000 rpm, 4 °C).
For protein purification, cell pellets were resuspended in lysis buffer (50 mM NaH2PO4, 400 mM NaCl, 1 mM TCEP, pH 7.5) and lysed by sonication. After removing cell debris by centrifugation, the lysate supernatant was purified by Ni-NTA resin with washing buffer (50 mM NaH2PO4, 150 mM NaCl, 100 mM imidazole, pH 7.5) and elution buffer (50 mM NaH2PO4, 150 mM NaCl, 300 mM imidazole, pH 7.5) and then dialyzed into the storage buffer (50 mM NaH2PO4, 150 mM NaCl, 1 mM TCEP, 10% glycerol, pH 7.5) and stored at −80 °C for further use. The P450 partner proteins, FdR and Fdx, were constructed according to the reported method [40].
For in vitro biochemical assays, the reaction mixture (50 µL) contained 0.4 mM substrate, 3 µM P450 enzyme, 5 µM FdR, 5 µM Fdx, 1 mM NADPH, 0.2 mM DTT, and 1 mM EDTA in 50 mM PBS buffer (pH 7.4). The reaction mixture was incubated at 37 °C for 1 h and quenched by adding 50 µL MeOH. After centrifugation (10 min, 13,000 rpm), the mixture was subjected to HPLC–UV or UHPLC–HRMS analysis.
Antioxidant activity
The antioxidant activity was assayed using a Total Antioxidant Capacity Assay Kit with ABTS method (Beyotime Biotechnology). Briefly, the fresh ABTS working solution was prepared by mixing ABTS stock solution with oxidant solution for an overnight reaction, and then this mixture was diluted 50 times by 80% ethanol. To initiate the assay reaction, 10 µL of each sample with a concentration of 1.0 mM in 80% ethanol was mixed with 200 µL of fresh ABTS working solution. After being incubated at 30 °C for 5 min, the 734 nm absorbance was measured on a Thermo Varioskan LUX Microplate reader. Trolox was used as the reference compound for calculation of ABTS-reducing activity (in %).
Computational ECD calculation of compound 1
Merck molecular force field (MMFF) and DFT as well as TDDFT calculations were carried out with the Spartan 14 software (Wavefunction Inc.) and the Gaussian 16 program [41], respectively. Conformers within the 2 kJ/mol energy window were generated and optimized using DFT calculations at the B3LYP/6-31+G (d,p) level. Frequency calculations were performed at the same level to confirm that each optimized conformer was true minimum and to estimate the relative thermal free energy (ΔG) at 298.15 K. The two conformers were chosen for ECD calculations in methanol at the B3LYP/6-311+G (d,p) level. Solvent effects were taken into consideration using the self-consistent reaction field (SCRF) method with the polarizable continuum model (PCM). The ECD spectrum was generated by the SpecDis program [42] using a Gaussian band shape with 0.23 eV exponential half-width from dipole length rotational strengths.
Supporting Information
| Supporting Information File 1: Detailed descriptions of the experimental procedures and comprehensive analytical data. | ||
| Format: PDF | Size: 6.0 MB | Download |
References
-
Liu, J.; Liu, A.; Hu, Y. Nat. Prod. Rep. 2021, 38, 1469–1505. doi:10.1039/d0np00063a
Return to citation in text: [1] [2] [3] -
Gigant, B.; Wang, C.; Ravelli, R. B. G.; Roussi, F.; Steinmetz, M. O.; Curmi, P. A.; Sobel, A.; Knossow, M. Nature 2005, 435, 519–522. doi:10.1038/nature03566
Return to citation in text: [1] -
Präg, A.; Grüning, B. A.; Häckh, M.; Lüdeke, S.; Wilde, M.; Luzhetskyy, A.; Richter, M.; Luzhetska, M.; Günther, S.; Müller, M. J. Am. Chem. Soc. 2014, 136, 6195–6198. doi:10.1021/ja501630w
Return to citation in text: [1] [2] -
Ma, J.; Wang, Z.; Huang, H.; Luo, M.; Zuo, D.; Wang, B.; Sun, A.; Cheng, Y.-Q.; Zhang, C.; Ju, J. Angew. Chem., Int. Ed. 2011, 50, 7797–7802. doi:10.1002/anie.201102305
Return to citation in text: [1] [2] -
Liu, X.-G.; Lu, X.; Gao, W.; Li, P.; Yang, H. Nat. Prod. Rep. 2022, 39, 474–511. doi:10.1039/d1np00026h
Return to citation in text: [1] -
Yin, L.; Liebscher, J. Chem. Rev. 2007, 107, 133–173. doi:10.1021/cr0505674
Return to citation in text: [1] -
Rahman, M.; Riaz, M.; Desai, U. R. Chem. Biodiversity 2007, 4, 2495–2527. doi:10.1002/cbdv.200790205
Return to citation in text: [1] -
Selepe, M. A.; Van Heerden, F. R. Molecules 2013, 18, 4739–4765. doi:10.3390/molecules18044739
Return to citation in text: [1] -
Shalit, H.; Dyadyuk, A.; Pappo, D. J. Org. Chem. 2019, 84, 1677–1686. doi:10.1021/acs.joc.8b03084
Return to citation in text: [1] -
Hüttel, W.; Müller, M. Nat. Prod. Rep. 2021, 38, 1011–1043. doi:10.1039/d0np00010h
Return to citation in text: [1] [2] [3] -
Hu, J.; Li, H.; Chooi, Y.-H. J. Am. Chem. Soc. 2019, 141, 8068–8072. doi:10.1021/jacs.9b03354
Return to citation in text: [1] -
Obermaier, S.; Thiele, W.; Fürtges, L.; Müller, M. Angew. Chem., Int. Ed. 2019, 58, 9125–9128. doi:10.1002/anie.201903759
Return to citation in text: [1] -
Agarwal, V.; El Gamal, A. A.; Yamanaka, K.; Poth, D.; Kersten, R. D.; Schorn, M.; Allen, E. E.; Moore, B. S. Nat. Chem. Biol. 2014, 10, 640–647. doi:10.1038/nchembio.1564
Return to citation in text: [1] [2] -
Mazzaferro, L. S.; Hüttel, W.; Fries, A.; Müller, M. J. Am. Chem. Soc. 2015, 137, 12289–12295. doi:10.1021/jacs.5b06776
Return to citation in text: [1] [2] -
Kudanga, T.; Nemadziva, B.; Le Roes-Hill, M. Appl. Microbiol. Biotechnol. 2017, 101, 13–33. doi:10.1007/s00253-016-7987-5
Return to citation in text: [1] -
Fürtges, L.; Obermaier, S.; Thiele, W.; Foegen, S.; Müller, M. ChemBioChem 2019, 20, 1928–1932. doi:10.1002/cbic.201900041
Return to citation in text: [1] -
Agarwal, V.; Moore, B. S. ACS Chem. Biol. 2014, 9, 1980–1984. doi:10.1021/cb5004338
Return to citation in text: [1] [2] -
Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7
Return to citation in text: [1] [2] [3] -
Guo, Z.; Li, P.; Chen, G.; Li, C.; Cao, Z.; Zhang, Y.; Ren, J.; Xiang, H.; Lin, S.; Ju, J.; Chen, Y. J. Am. Chem. Soc. 2018, 140, 18009–18015. doi:10.1021/jacs.8b10136
Return to citation in text: [1] [2] -
Ferreira, D.; Bekker, R. Nat. Prod. Rep. 1996, 13, 411–433. doi:10.1039/np9961300411
Return to citation in text: [1] -
Sohn, S. I.; Pandian, S.; Oh, Y. J.; Kang, H. J.; Cho, W. S.; Cho, Y. S. Front. Plant Sci. 2021, 12, 670103. doi:10.3389/fpls.2021.670103
Return to citation in text: [1] -
Al-Maharik, N. Nat. Prod. Rep. 2019, 36, 1156–1195. doi:10.1039/c8np00069g
Return to citation in text: [1] -
Lewis, D. F. V.; Wiseman, A. Enzyme Microb. Technol. 2005, 36, 377–384. doi:10.1016/j.enzmictec.2004.07.018
Return to citation in text: [1] -
Chun, Y. J.; Kim, M. Y.; Guengerich, F. P. Biochem. Biophys. Res. Commun. 1999, 262, 20–24. doi:10.1006/bbrc.1999.1152
Return to citation in text: [1] -
Funa, N.; Funabashi, M.; Ohnishi, Y.; Horinouchi, S. J. Bacteriol. 2005, 187, 8149–8155. doi:10.1128/jb.187.23.8149-8155.2005
Return to citation in text: [1] [2] -
Zhao, B.; Guengerich, F. P.; Voehler, M.; Waterman, M. R. J. Biol. Chem. 2005, 280, 42188–42197. doi:10.1074/jbc.m509220200
Return to citation in text: [1] -
Zhao, B.; Lamb, D. C.; Lei, L.; Kelly, S. L.; Yuan, H.; Hachey, D. L.; Waterman, M. R. Biochemistry 2007, 46, 8725–8733. doi:10.1021/bi7006959
Return to citation in text: [1] -
Kraus, P. F.; Kutchan, T. M. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2071–2075. doi:10.1073/pnas.92.6.2071
Return to citation in text: [1] -
Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C. T.; Tang, Y. Chem. Rev. 2017, 117, 5226–5333. doi:10.1021/acs.chemrev.6b00478
Return to citation in text: [1] -
Funa, N.; Ohnishi, Y.; Fujii, I.; Shibuya, M.; Ebizuka, Y.; Horinouchi, S. Nature 1999, 400, 897–899. doi:10.1038/23748
Return to citation in text: [1] -
Álvarez-Álvarez, R.; Botas, A.; Albillos, S. M.; Rumbero, A.; Martín, J. F.; Liras, P. Microb. Cell Fact. 2015, 14, 178. doi:10.1186/s12934-015-0373-7
Return to citation in text: [1] -
Lim, Y.-R.; Han, S.; Kim, J.-H.; Park, H.-G.; Lee, G.-Y.; Le, T.-K.; Yun, C.-H.; Kim, D. Biomol. Ther. 2017, 25, 171–176. doi:10.4062/biomolther.2016.182
Return to citation in text: [1] -
Ungar, Y.; Osundahunsi, O. F.; Shimoni, E. J. Agric. Food Chem. 2003, 51, 4394–4399. doi:10.1021/jf034021z
Return to citation in text: [1] -
Heim, K. E.; Tagliaferro, A. R.; Bobilya, D. J. J. Nutr. Biochem. 2002, 13, 572–584. doi:10.1016/s0955-2863(02)00208-5
Return to citation in text: [1] -
Weston, L. A.; Mathesius, U. J. Chem. Ecol. 2013, 39, 283–297. doi:10.1007/s10886-013-0248-5
Return to citation in text: [1] -
Sugiyama, A. J. Adv. Res. 2019, 19, 67–73. doi:10.1016/j.jare.2019.03.005
Return to citation in text: [1] -
Katoh, K.; Standley, D. M. Mol. Biol. Evol. 2013, 30, 772–780. doi:10.1093/molbev/mst010
Return to citation in text: [1] -
Price, M. N.; Dehal, P. S.; Arkin, A. P. PLoS One 2010, 5, e9490. doi:10.1371/journal.pone.0009490
Return to citation in text: [1] -
Xia, Y.; Li, K.; Li, J.; Wang, T.; Gu, L.; Xun, L. Nucleic Acids Res. 2019, 47, e15. doi:10.1093/nar/gky1169
Return to citation in text: [1] -
Zhang, W.; Du, L.; Li, F.; Zhang, X.; Qu, Z.; Han, L.; Li, Z.; Sun, J.; Qi, F.; Yao, Q.; Sun, Y.; Geng, C.; Li, S. ACS Catal. 2018, 8, 9992–10003. doi:10.1021/acscatal.8b02913
Return to citation in text: [1] -
Gaussian 16, Rev. A.03; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] -
Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. Chirality 2013, 25, 243–249. doi:10.1002/chir.22138
Return to citation in text: [1]
| 38. | Price, M. N.; Dehal, P. S.; Arkin, A. P. PLoS One 2010, 5, e9490. doi:10.1371/journal.pone.0009490 |
| 39. | Xia, Y.; Li, K.; Li, J.; Wang, T.; Gu, L.; Xun, L. Nucleic Acids Res. 2019, 47, e15. doi:10.1093/nar/gky1169 |
| 40. | Zhang, W.; Du, L.; Li, F.; Zhang, X.; Qu, Z.; Han, L.; Li, Z.; Sun, J.; Qi, F.; Yao, Q.; Sun, Y.; Geng, C.; Li, S. ACS Catal. 2018, 8, 9992–10003. doi:10.1021/acscatal.8b02913 |
| 1. | Liu, J.; Liu, A.; Hu, Y. Nat. Prod. Rep. 2021, 38, 1469–1505. doi:10.1039/d0np00063a |
| 5. | Liu, X.-G.; Lu, X.; Gao, W.; Li, P.; Yang, H. Nat. Prod. Rep. 2022, 39, 474–511. doi:10.1039/d1np00026h |
| 3. | Präg, A.; Grüning, B. A.; Häckh, M.; Lüdeke, S.; Wilde, M.; Luzhetskyy, A.; Richter, M.; Luzhetska, M.; Günther, S.; Müller, M. J. Am. Chem. Soc. 2014, 136, 6195–6198. doi:10.1021/ja501630w |
| 4. | Ma, J.; Wang, Z.; Huang, H.; Luo, M.; Zuo, D.; Wang, B.; Sun, A.; Cheng, Y.-Q.; Zhang, C.; Ju, J. Angew. Chem., Int. Ed. 2011, 50, 7797–7802. doi:10.1002/anie.201102305 |
| 4. | Ma, J.; Wang, Z.; Huang, H.; Luo, M.; Zuo, D.; Wang, B.; Sun, A.; Cheng, Y.-Q.; Zhang, C.; Ju, J. Angew. Chem., Int. Ed. 2011, 50, 7797–7802. doi:10.1002/anie.201102305 |
| 18. | Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7 |
| 19. | Guo, Z.; Li, P.; Chen, G.; Li, C.; Cao, Z.; Zhang, Y.; Ren, J.; Xiang, H.; Lin, S.; Ju, J.; Chen, Y. J. Am. Chem. Soc. 2018, 140, 18009–18015. doi:10.1021/jacs.8b10136 |
| 3. | Präg, A.; Grüning, B. A.; Häckh, M.; Lüdeke, S.; Wilde, M.; Luzhetskyy, A.; Richter, M.; Luzhetska, M.; Günther, S.; Müller, M. J. Am. Chem. Soc. 2014, 136, 6195–6198. doi:10.1021/ja501630w |
| 23. | Lewis, D. F. V.; Wiseman, A. Enzyme Microb. Technol. 2005, 36, 377–384. doi:10.1016/j.enzmictec.2004.07.018 |
| 24. | Chun, Y. J.; Kim, M. Y.; Guengerich, F. P. Biochem. Biophys. Res. Commun. 1999, 262, 20–24. doi:10.1006/bbrc.1999.1152 |
| 2. | Gigant, B.; Wang, C.; Ravelli, R. B. G.; Roussi, F.; Steinmetz, M. O.; Curmi, P. A.; Sobel, A.; Knossow, M. Nature 2005, 435, 519–522. doi:10.1038/nature03566 |
| 25. | Funa, N.; Funabashi, M.; Ohnishi, Y.; Horinouchi, S. J. Bacteriol. 2005, 187, 8149–8155. doi:10.1128/jb.187.23.8149-8155.2005 |
| 26. | Zhao, B.; Guengerich, F. P.; Voehler, M.; Waterman, M. R. J. Biol. Chem. 2005, 280, 42188–42197. doi:10.1074/jbc.m509220200 |
| 27. | Zhao, B.; Lamb, D. C.; Lei, L.; Kelly, S. L.; Yuan, H.; Hachey, D. L.; Waterman, M. R. Biochemistry 2007, 46, 8725–8733. doi:10.1021/bi7006959 |
| 17. | Agarwal, V.; Moore, B. S. ACS Chem. Biol. 2014, 9, 1980–1984. doi:10.1021/cb5004338 |
| 18. | Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7 |
| 19. | Guo, Z.; Li, P.; Chen, G.; Li, C.; Cao, Z.; Zhang, Y.; Ren, J.; Xiang, H.; Lin, S.; Ju, J.; Chen, Y. J. Am. Chem. Soc. 2018, 140, 18009–18015. doi:10.1021/jacs.8b10136 |
| 21. | Sohn, S. I.; Pandian, S.; Oh, Y. J.; Kang, H. J.; Cho, W. S.; Cho, Y. S. Front. Plant Sci. 2021, 12, 670103. doi:10.3389/fpls.2021.670103 |
| 10. | Hüttel, W.; Müller, M. Nat. Prod. Rep. 2021, 38, 1011–1043. doi:10.1039/d0np00010h |
| 15. | Kudanga, T.; Nemadziva, B.; Le Roes-Hill, M. Appl. Microbiol. Biotechnol. 2017, 101, 13–33. doi:10.1007/s00253-016-7987-5 |
| 16. | Fürtges, L.; Obermaier, S.; Thiele, W.; Foegen, S.; Müller, M. ChemBioChem 2019, 20, 1928–1932. doi:10.1002/cbic.201900041 |
| 1. | Liu, J.; Liu, A.; Hu, Y. Nat. Prod. Rep. 2021, 38, 1469–1505. doi:10.1039/d0np00063a |
| 10. | Hüttel, W.; Müller, M. Nat. Prod. Rep. 2021, 38, 1011–1043. doi:10.1039/d0np00010h |
| 11. | Hu, J.; Li, H.; Chooi, Y.-H. J. Am. Chem. Soc. 2019, 141, 8068–8072. doi:10.1021/jacs.9b03354 |
| 12. | Obermaier, S.; Thiele, W.; Fürtges, L.; Müller, M. Angew. Chem., Int. Ed. 2019, 58, 9125–9128. doi:10.1002/anie.201903759 |
| 13. | Agarwal, V.; El Gamal, A. A.; Yamanaka, K.; Poth, D.; Kersten, R. D.; Schorn, M.; Allen, E. E.; Moore, B. S. Nat. Chem. Biol. 2014, 10, 640–647. doi:10.1038/nchembio.1564 |
| 14. | Mazzaferro, L. S.; Hüttel, W.; Fries, A.; Müller, M. J. Am. Chem. Soc. 2015, 137, 12289–12295. doi:10.1021/jacs.5b06776 |
| 6. | Yin, L.; Liebscher, J. Chem. Rev. 2007, 107, 133–173. doi:10.1021/cr0505674 |
| 7. | Rahman, M.; Riaz, M.; Desai, U. R. Chem. Biodiversity 2007, 4, 2495–2527. doi:10.1002/cbdv.200790205 |
| 8. | Selepe, M. A.; Van Heerden, F. R. Molecules 2013, 18, 4739–4765. doi:10.3390/molecules18044739 |
| 9. | Shalit, H.; Dyadyuk, A.; Pappo, D. J. Org. Chem. 2019, 84, 1677–1686. doi:10.1021/acs.joc.8b03084 |
| 20. | Ferreira, D.; Bekker, R. Nat. Prod. Rep. 1996, 13, 411–433. doi:10.1039/np9961300411 |
| 42. | Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. Chirality 2013, 25, 243–249. doi:10.1002/chir.22138 |
| 14. | Mazzaferro, L. S.; Hüttel, W.; Fries, A.; Müller, M. J. Am. Chem. Soc. 2015, 137, 12289–12295. doi:10.1021/jacs.5b06776 |
| 18. | Zetzsche, L. E.; Yazarians, J. A.; Chakrabarty, S.; Hinze, M. E.; Murray, L. A. M.; Lukowski, A. L.; Joyce, L. A.; Narayan, A. R. H. Nature 2022, 603, 79–85. doi:10.1038/s41586-021-04365-7 |
| 13. | Agarwal, V.; El Gamal, A. A.; Yamanaka, K.; Poth, D.; Kersten, R. D.; Schorn, M.; Allen, E. E.; Moore, B. S. Nat. Chem. Biol. 2014, 10, 640–647. doi:10.1038/nchembio.1564 |
| 17. | Agarwal, V.; Moore, B. S. ACS Chem. Biol. 2014, 9, 1980–1984. doi:10.1021/cb5004338 |
| 28. | Kraus, P. F.; Kutchan, T. M. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2071–2075. doi:10.1073/pnas.92.6.2071 |
| 35. | Weston, L. A.; Mathesius, U. J. Chem. Ecol. 2013, 39, 283–297. doi:10.1007/s10886-013-0248-5 |
| 36. | Sugiyama, A. J. Adv. Res. 2019, 19, 67–73. doi:10.1016/j.jare.2019.03.005 |
| 37. | Katoh, K.; Standley, D. M. Mol. Biol. Evol. 2013, 30, 772–780. doi:10.1093/molbev/mst010 |
| 33. | Ungar, Y.; Osundahunsi, O. F.; Shimoni, E. J. Agric. Food Chem. 2003, 51, 4394–4399. doi:10.1021/jf034021z |
| 34. | Heim, K. E.; Tagliaferro, A. R.; Bobilya, D. J. J. Nutr. Biochem. 2002, 13, 572–584. doi:10.1016/s0955-2863(02)00208-5 |
| 31. | Álvarez-Álvarez, R.; Botas, A.; Albillos, S. M.; Rumbero, A.; Martín, J. F.; Liras, P. Microb. Cell Fact. 2015, 14, 178. doi:10.1186/s12934-015-0373-7 |
| 25. | Funa, N.; Funabashi, M.; Ohnishi, Y.; Horinouchi, S. J. Bacteriol. 2005, 187, 8149–8155. doi:10.1128/jb.187.23.8149-8155.2005 |
| 32. | Lim, Y.-R.; Han, S.; Kim, J.-H.; Park, H.-G.; Lee, G.-Y.; Le, T.-K.; Yun, C.-H.; Kim, D. Biomol. Ther. 2017, 25, 171–176. doi:10.4062/biomolther.2016.182 |
| 1. | Liu, J.; Liu, A.; Hu, Y. Nat. Prod. Rep. 2021, 38, 1469–1505. doi:10.1039/d0np00063a |
| 10. | Hüttel, W.; Müller, M. Nat. Prod. Rep. 2021, 38, 1011–1043. doi:10.1039/d0np00010h |
| 29. | Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C. T.; Tang, Y. Chem. Rev. 2017, 117, 5226–5333. doi:10.1021/acs.chemrev.6b00478 |
| 30. | Funa, N.; Ohnishi, Y.; Fujii, I.; Shibuya, M.; Ebizuka, Y.; Horinouchi, S. Nature 1999, 400, 897–899. doi:10.1038/23748 |
© 2022 Liu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.