Abstract
This article describes the detailed analysis of the reaction between arylamines, such as aniline, o-anisidine, and methyl anthranilate, with selenium dioxide in acetonitrile. A systematic analysis of the reaction products with the help of 77Se NMR and single-crystal X-ray crystallography revealed that the reaction progress follows three major reaction pathways, electrophilic selenation, oxidative polymerization, and solvent oxidation. For aniline and o-anisidine, predominant oxidative polymerization occurred, leading to the formation of the respective polyaniline polymers as major products. For methyl anthranilate, the oxidative polymerization was suppressed due to the delocalization of amine lone pair electrons over the adjacent carboxylate function, which prompted the selenation pathway, leading to the formation of two of the isomeric diorganyl selenides of methyl anthranilate. The diaryl selenides were structurally characterized using single-crystal X-ray diffraction. Density functional theory calculations suggest that the highest occupied molecular orbital of methyl anthranilate was deeply buried, which suppressed the oxidative polymerization pathway. Due to solvent oxidation, oxamide formation was also noticed to a considerable extent. This study provides that utmost care must be exercised while using SeO2 as an electrophile source in aromatic electrophilic substitution reactions.

Graphical Abstract
Introduction
Organoselenium compounds have received considerable attention due to interesting medicinal properties, such as antioxidative [1,2], antimicrobial [3,4], and anticancer activity [5,6]. Several classes of organoselenium compounds are known to imitate the glutathione peroxidase [7-11]. Consequently, the development of new strategies for selenation of organic compounds has attracted considerable interest [12,13]. The various approaches used for selenation of aromatic compounds include directed lithiation [14,15], copper-catalyzed selenation [16-18], and aromatic nucleophilic substitution reactions [19-22]. Electrophilic selenium reagents (e.g., phenylselenenyl bromide) have often been used in oxyselenenylation of olefins, which follows an electrophilic addition mechanism [23-25]. However, such reagents are rarely used for electrophilic substitution of aromatic systems. Recently, notable progress has been made in the use of aromatic electrophilic substitution to synthesize arylselenium compounds [26-28]. Noteworthy examples are the use of SeO2 as selenium source in aromatic electrophilic substitution reactions [27-29]. Selenium dioxide is a well-known oxidizing agent for the allylic oxidation and oxidation of α-CH bonds located adjacent to electron-withdrawing groups [30-32]. Due to the oxidative nature, the use as selenium source in the synthesis of arylchalcogen compounds, in particular diorganyl selenides, can be a challenging process that involves sequential formation of two C–Se bonds and reduction of a Se=O bond in a single-pot reaction. Further, regioselective aromatic electrophilic substitution is often difficult. Various synthetic strategies have evolved to address such problems and expand the scope of SeO2 beyond the oxidizing capability. Ren et al. adopted potassium-iodide-mediated catalytic selenation of aromatic compounds using SeO2 (Scheme 1) [33]. This reaction comprises four main steps: (i) iodide-mediated aryl transfer from boronic acid to selenium dioxide, (ii) reduction of arylseleninic acid to diaryl diselenide, (iii) oxidation of diaryl diselenide to aryl selenenyl iodide with iodine, and (iv) electrophilic substitution of aniline derivatives. With this approach, electrophilic substitution can be achieved in ortho and para positions of aniline derivatives by careful selection of the substrates. Kumar et al. used SeO2 and phenylboronic acid to make symmetrical diaryl selenides [34]. In both cases, aryl transfer from the boron to the selenium center appeared to play a crucial role in the formation of the C–Se bond. AlCl3-catalyzed formation of benzoselenazole using SeO2 as electrophilic source has also been reported [35]. Quell et al. reported the syntheses of diaryl selenides and biphenol derivatives using SeO2 and phenols with one position (ortho or para) available for electrophilic substitution [36]. They found that solvent played a significant role in directing the outcome of the reaction. In protic solvents, biphenols were selectively formed through C–C bond formation, whereas in pyridine, the generation of diaryl selenide derivatives was almost exclusively promoted via aromatic electrophilic substitution. All of these reactions reveal the importance of using catalytic processes, preactivated substrates, or of blocking ortho or para sites to obtain the desired arylchalcogen compounds in good yield.
![[1860-5397-20-105-i1]](/bjoc/content/inline/1860-5397-20-105-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reported synthetic methods for the selenation of aromatic compounds.
Scheme 1: Reported synthetic methods for the selenation of aromatic compounds.
To our surprise, Bhat et al. have very recently reported the synthesis of the black solid 4,4'-selenodianiline without the use of catalysts, preactivation, or any blocking groups (Scheme 1) [37]. In contrast, Kim et al., utilizing a CuI-catalyzed reaction [38], have reported that 4,4'-selenodianiline is a pale brown solid, which conforms to our previous experience that diaryl selenides and diaryl diselenides are frequently yellow to brownish orange solids or liquids [39,40]. These contradicting observations prompted us to reinvestigate the reaction reported by Bhat et al. in detail. Indeed, we noticed that the reaction of SeO2 with arylamines follows a complex reaction pathway, leading to a mixture of compounds. We established the possible reaction pathways using 77Se NMR spectroscopy and single-crystal X-ray crystallographic studies. Density functional theory (DFT) calculations were carried out to understand the relative polymerization tendency of aniline, o-anisidine, and methyl anthranilate in the presence of SeO2.
Results and Discussion
Reaction of aniline with SeO2
The reaction of aniline with SeO2 was carried out in acetonitrile in a nitrogen atmosphere (Scheme 2). It is worth mentioning that while the number of moles of reactants employed in our experiment was slightly different as compared to the condition mentioned in the work of Bhat. et. al [37], similar results were obtained under both conditions. Specifically, under our conditions, the amount of reactant used was 10 times higher than that used by Bhat. et. al., and the amount of solvent was doubled. We used a higher concentration of reactants in our studies as the yield of soluble products obtained at lower concentration of reactants was not satisfactory. However, under both conditions, a blackish purple polymeric solid was isolated as major product, labelled polymer 1. The polymer was isolated from the reaction after 3 h, and the supernatant was analyzed for other soluble products. The UV–vis spectrum of polymer 1 dispersed in methanol showed two major peaks at ≈271 and ≈561 nm (Figure S1, Supporting Information File 1), corresponding to the π→π* and benzenoid→quinonoid excitonic transitions, respectively, which was characteristic of polyaniline existing as emeraldine free base [41,42]. The polyaniline nature was further confirmed by FTIR spectroscopy (Figure S2, Supporting Information File 1). The broad peak observed in the range of 3300–3400 cm−1 was due to N–H stretching of the polymer [43,44]. The peak appearing at around 3000 cm−1 corresponded to the aryl C–H stretching. The peaks observed at 1593 and 1479 cm−1 were associated with C=C and C=N stretching, respectively. The obtained result was further supported by solid-state synthesis of selenious acid containing polyaniline via chemical oxidation of aniline with SeO2 as oxidant [45]. Further, Tanini et al. described that selenium dioxide has potential to oxidize aniline to form nitrobenzene in aqueous medium [46]. These findings irrevocably confirmed that aniline undergoes significant oxidative polymerization in the presence of SeO2.
![[1860-5397-20-105-i2]](/bjoc/content/inline/1860-5397-20-105-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of selenium dioxide with aniline.
Scheme 2: Reaction of selenium dioxide with aniline.
After recognizing the polymeric nature of the solid, we analyzed the small molecular species present in the supernatant. Thin-layer chromatography (TLC) of the supernatant revealed that it contained a mixture of compounds. We could not isolate monoselenide 1 as a pure compound through column chromatography. The obtained fraction was a black solid as reported by Bhat et al., and HRMS analysis revealed a peak at m/z 265.0229, corresponding to [M + H]+ (Figure S4, Supporting Information File 1). However, we could not ascertain the identity of the compound based on HRMS data. To test whether the isolated black solid was a single compound or an isomeric mixture of compounds 1 and 2, 77Se NMR spectroscopy could be a convenient tool. Unfortunately, 77Se NMR data was not reported in the work of Bhat et al. [37]. With this question in mind, the reaction was repeated several times to obtain a sufficient amount of the mixture for 77Se NMR spectroscopy. Indeed, two major resonance signals at 371 and 296 ppm were observed in the 77Se NMR spectrum of the mixture recorded in DMSO-d6 (Figure S5, Supporting Information File 1), which could be assigned to diaryl monoselenides 1 and 2, respectively. The signal at 371 ppm corresponded to compound 1, based on a previous report [47]. In turn, the other signal was assigned to the isomeric compound 2. A similar observation was made in the 77Se NMR spectra of the pure diaryl diselenide isomers obtained from the reaction of methyl anthranilate with SeO2 (vide infra), which was also supported by single-crystal X-ray analysis. However, for aniline, in addition to the major polymerization and poor selenation process, solvent oxidation was also noted. Importantly, through column chromatography, the oxamide derivative 3 formed from through solvent (acetonitrile) oxidation was isolated as a colorless solid (vide infra). It was characterized by FTIR, 1H NMR, and mass spectrometry. The FTIR and mass spectrometry data were in accordance with the literature report [48,49]. Interestingly, the mass spectrum of the oxide 3 showed a signal at m/z 518.8590, corresponding to the 2:1 potassium complex [2M + K]+ (Figure S7, Supporting Information File 1).
Reaction of o-anisidine with SeO2
To further understand the competitive process, we extended the reaction to o-anisidine with an electron-donating methoxy group in an ortho position relative to NH2. Scheme 3 shows the reaction of o-anisidine with SeO2. Similar to aniline, a large quantity of o-anisidine was transformed into polymer, labelled polymer 2. The UV–vis and FTIR spectra of polymer 2 are shown in Figures S1 and S2, Supporting Information File 1, respectively. The spectral characteristics were consistent with those of the emeraldine free base, as discussed above for polymer 1.
![[1860-5397-20-105-i3]](/bjoc/content/inline/1860-5397-20-105-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of selenium dioxide with o-anisidine.
Scheme 3: Reaction of selenium dioxide with o-anisidine.
In the next step, the supernatant was analyzed by TLC and subsequently purified by column chromatography. Attempts to isolate the organoselenium compounds were unsuccessful due to the similar retention factor and low quantity. Therefore, all fractions were combined and subjected to 77Se NMR spectroscopy. The 77Se NMR spectrum of the mixture showed resonance signals at 406, 422, 537, 568, and 793 ppm (Figure S11, Supporting Information File 1). The peaks observed in the shielded region at 406 and 422 ppm were assigned to the unsymmetrical and symmetrical diaryl monoselenides 4 and 5. But it was unclear which peak corresponded to which. Similarly, the peaks at 537 and 568 ppm could be tentatively assigned to diaryl diselenides 6 and 7. The peak observed in the most deshielded region at 793 ppm was generally attributed of diaryl selenoxides such as compound 8 [39]. Despite the complexity, we could confirm the [M + H]+ ions of the isomeric monoselenides 4 and 5 from HRMS data (Figure S10, Supporting Information File 1). In addition to the above mentioned (possible) organoselenium compounds, the oxamide 9 and the quinone derivative 2,5-bis((2-methoxyphenyl)amino)cyclohexa-2,5-diene-1,4-dione (10) were isolated from the mixture. The spectroscopic data were consistent with earlier reports on the direct synthesis of 9 [50,51] and 10 [52]. The mass spectrum of oxamide 9 showed peaks corresponding to the sodium complexes [M + Na]+, [2M + Na]+, and [3M + Na]+ (Figure S13, Supporting Information File 1). The mass spectrum of quinone 10 showed a peak for [M + H]+ (Figure S17, Supporting Information File 1).
Reaction of methyl anthranilate with SeO2
After observing that unsubstituted aniline and aniline with an electron-donating function (i.e., o-anisidine) undergo significant polymerization in the presence of SeO2, we were interested in studying this reaction using methyl anthranilate, having an electron-withdrawing group (Scheme 4). To our surprise, the quantity of polymer 3 formed was low. After isolating ≈360 mg of the semisolid blackish dark polymer 3, the supernatant was subjected to column chromatography, which afforded three compounds, namely diaryl monoselenides 11 (87 mg, 4.6%) and 12 (≈476 mg, 25%) as well as oxamide 13 (2.2 mg, 0.12%). Polymer 3 was characterized using UV and FTIR spectroscopy (Figures S1 and S2, Supporting Information File 1). The spectral data were consistent with the emeraldine free base. Further, a characteristic n→π* transition for the CO group was noticed in the UV spectrum of polymer 3, which was absent in the UV spectra of the other two polymers [53]. The mass spectra of purified compounds 11 and 12 showed peaks for [M + H]+ (Figures S21 and S26, Supporting Information File 1). The 77Se NMR spectra of the purified isomers 11 and 12 showed resonance signals at 300 and 394 ppm, respectively (Figures S24 and S29, Supporting Information File 1). Similar to the 77Se NMR signals of isomeric diaryl diselenides 1 and 2, the symmetrical diaryl diselenide 12, having an amino group para to the selenium center in both aryl units, showed a downfield shift compared to isomer 11. The mass spectrum of oxamide 13 revealed peaks for the sodium complexes [M + Na]+, [2M + Na]+, and [3M + Na]+. Spectroscopic data of this compound, including FTIR as well as 1H and 13C NMR, matched those reported earlier [54].
![[1860-5397-20-105-i4]](/bjoc/content/inline/1860-5397-20-105-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of methyl anthranilate with SeO2.
Scheme 4: Reaction of methyl anthranilate with SeO2.
Relative extent of polymerization
To compare the relative polymerization tendency, we conducted reactions of arylamines with SeO2 in acetonitrile in sample vials and monitored the apparent color change (Figure S34, Supporting Information File 1). Instantaneous color change from colorless to blackish dark was observed for an o-anisidine solution after addition of SeO2. For an aniline solution, the color became light pink immediately upon addition of SeO2. For a methyl anthranilate solution, no color change was observed immediately upon addition of SeO2 as well as after 5 min of heating. The color of aniline and o-anisidine solutions became very intense after 5 min of heating. After 5 min of heating, the color intensity followed the following order: o-anisidine > aniline > methyl anthranilate. This clearly indicated that the relative extent of polymer formation decreased in the following order: o-anisidine > aniline > methyl anthranilate. As a consequence of the poor polymerization tendency, polymer 3 was obtained in a smaller amount of ≈360 mg after 24 h. Whereas at the same molar ratio, polymers 1 and 2 were obtained in larger quantities of ≈2.3 g after the short time of 3 h (see Experimental section for details).
Mechanistic aspects
Mechanism for the formation of diaryl monoselenides
The plausible mechanism for the formation of diaryl monoselenides is shown in Scheme 5. The first step is the electrophilic substitution of SeO2 on the aromatic ring, either in the ortho or para position. The resulting arylseleninic acid acts as an electrophile, substituting a proton in another aniline molecule, leading to a hydrated selenoxide. This can give rise to either diaryl selenoxide via dehydration or diaryl monoselenide via reductive elimination by eliminating H2O2 [39]. Observation of m/z peaks for compound 8 clearly confirmed the formation of diaryl selenoxide in the reaction.
![[1860-5397-20-105-i5]](/bjoc/content/inline/1860-5397-20-105-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction mechanism for the formation of diaryl monoselenides.
Scheme 5: Reaction mechanism for the formation of diaryl monoselenides.
Mechanism for the formation of oxamides
The possible reaction mechanism for the formation of oxamide is shown in Scheme 6. Formation of acetanilide in the reaction of aniline and acetonitrile is known to occur in the presence of Lewis acid catalyst Al2O3 [55]. In our case, either SeO2 (Lewis acid) or H2SeO3 (Brønsted acid) may act as acid catalyst to convert aniline into acetanilide using acetonitrile as acetylating agent. The resulting acetanilide underwent α-CH oxidation with SeO2 to give 2-oxo-N-phenylacetamide, which undergoes Schiff base formation with aniline. The resulting Schiff base undergoes further α-CH oxidation with SeO2 to give the oxamide 3. A similar mechanism may be assumed for the formation of the other oxamides 9 and 13.
![[1860-5397-20-105-i6]](/bjoc/content/inline/1860-5397-20-105-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction mechanism for the formation of oxamides.
Scheme 6: Reaction mechanism for the formation of oxamides.
Mechanism for the formation of 2,5-bis((2-methoxyphenyl)amino)cyclohexa-2,5-diene-1,4-dione (10)
Generally, formation of polyaniline occurs through a radical mechanism. Such a radical mechanism is relevant for the formation of quinones having exceptional radical-stabilizing abilities. The best example in nature is the radical pathway in the catechol oxidation process [56-58]. The structure of o-anisidine resembles catechol as it has two adjacent electron-donating functions (NH2 and OMe). For o-anisidine, the amine radical resulting from reaction of o-anisidine with SeO2 is stabilized by resonance (Scheme 7). It combines with the hydroxyl radical and undergoes subsequent oxidation to give 2-methoxycyclohexa-2,5-diene-1,4-dione. This iminoquinone upon hydrolysis gives 2-methoxycyclohexa-2,5-diene-1,4-dione, and the latter undergoes addition–elimination reaction with another molecule of o-anisidine to give 2-(phenylamino)cyclohexa-2,5-diene-1,4-dione. The third unit of o-anisidine is added to the quinone via 1,4-addition, followed by oxidation of the 1,4-dihydroxy compound to give 2,5-bis((2-methoxyphenyl)amino)cyclohexa-2,5-diene-1,4-dione (compound 10).
![[1860-5397-20-105-i7]](/bjoc/content/inline/1860-5397-20-105-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction mechanism for the formation of quinone 10.
Scheme 7: Reaction mechanism for the formation of quinone 10.
Single-crystal X-ray crystallographic studies
The ORTEP diagram of oxamide 3 is shown in Figure 1. Compound 3 crystallized in a monoclinic crystal system in the P21/n space group. The single-crystal X-ray structure of compound 3 was reported with the space group P21/c [49]. It adopts a transoid geometry around the oxamide C–C bond with nearly 180° torsion angle. This provides the molecule with a planar geometry. It shows intermolecular hydrogen bonding between the amide O and NH moieties. (Figure S35, Supporting Information File 1).
![[1860-5397-20-105-1]](/bjoc/content/figures/1860-5397-20-105-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 3. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): O(1)–C(7) 1.2207 (16), N(1)–C(7) 1.3356 (17), N(1)–C(1) 1.4193 (16), N(1)–H(1) 0.8600.
Figure 1: Molecular structure of 3. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): O...
Oxamide 9 crystallized in a monoclinic crystal system in the space group P21/c. The structure was similar to the structure of oxamide 3 (Figure 2). The hydrogen of the NH unit was concealed by the oxygen atoms of the C=O and OMe units. Consequently, it did not engage in intermolecular hydrogen bonding. Due to the planar structure, it self-organized through π–π interaction between the oxamide motif and the aryl π-framework. Further, the CH…O=C interaction facilitated the formation of ladder-like packing in the solid state (Figure S36, Supporting Information File 1).
![[1860-5397-20-105-2]](/bjoc/content/figures/1860-5397-20-105-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of 9. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): O(1)–C(7) 1.2192 (18), N(1)–C(7) 1.3396, N(1)–C(1) 1.4050 (19), N(1)–H(1) 0.8600, O(2)–C(2) 1.3674 (18), O(2)–C(8) 1.420 (2).
Figure 2: Molecular structure of 9. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): O...
The molecular structure of oxamide 13 is shown in Figure 3. It crystallized in a triclinic crystal system in the space group P−1. It was structurally similar to the other oxamides 3 and 9. It showed an intramolecular hydrogen bonding between the NH and the carbonyl group of the methoxy ester. A layer-by-layer packing was observed in the crystal structure (Figure S37, Supporting Information File 1). The dipolar characteristics of the ester group appeared to assist the molecular layer-by-layer stacking seen in the crystal packing.
![[1860-5397-20-105-3]](/bjoc/content/figures/1860-5397-20-105-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of 13. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): O(3)–C(7) 1.2070 (16), O(1)–C(7) 1.3322 (16), N(1)–H(1) 0.8600, N(1)–C(6) 1.4044 (16), N(1)–C(9) 1.3440 (16), O(2)–C(9) 1.2160 (16).
Figure 3: Molecular structure of 13. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å): ...
The molecular structure of the quinone derivative 10 is shown in Figure 4. It crystallized in a monoclinic crystal system in the space group C2/c.
![[1860-5397-20-105-4]](/bjoc/content/figures/1860-5397-20-105-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure of 10. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å) and angles (°): O(1)–C(2) 1.2300 (16), N(1)–C(1) 1.3461 (17), N(1)–H(1) 0.884 (19), N(1)–C(4) 1.4078 (18), O(2)–C(5) 1.3610 (17), O(2)–C(10) 1.4308 (18), C(5)–O(2)–C(10) 118.06, C(1)–N(1)–C(4) 128.39 (12).
Figure 4: Molecular structure of 10. Thermal ellipsoids drawn at 50% probability. Selected bond lengths (Å) a...
The molecular structure of the diorganyl monoselenide 11 is shown in Figure 5. It crystallized in a triclinic crystal system in the space group P−1. The C–Se–C bond angle was found to be 99.01°. Both inter- and intramolecular hydrogen bonding were noted in the structure (Figure S39, Supporting Information File 1). It allowed the molecule to self-align in a repeating cyclic ring pattern in one dimension.
![[1860-5397-20-105-5]](/bjoc/content/figures/1860-5397-20-105-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure of 11. Thermal ellipsoids drawn at 50% probability. Selected bond angles (°): C(7)–Se(1)–C(1) 99.01 (8), N(1)–C(2)–C(1) 120.34 (18), N(1)–C(2)–C(3) 122.05 (18), O(3)–C(13)–O(1) 121.5(2), O(3)–C(13)–C(3) 125.4 (2), N(2)–C(10)–C(9) 123.04 (19), O(4)–C(15)–O(2), O(4)–C(15)–O(2) 121.5 (2), O(4)–C(15)–C(9) 125.4 (2).
Figure 5: Molecular structure of 11. Thermal ellipsoids drawn at 50% probability. Selected bond angles (°): C...
The molecular structure of the diorganyl monoselenide 12 is shown in Figure 6. It crystallized in an orthorhombic crystal system in the space group P21212. The C–Se–C bond angle was 100.11°. Since the NH2 groups in this molecule were further away from each other compared to the monoselenide 11, the hydrogen bonding arrangement led to a two-dimensional packing with a repeating zigzag pattern. Curiously, this compound showed an unusual Se···C σ-hole–π chalcogen bonding. Such interactions are rarely reported and currently gaining interest [59].
![[1860-5397-20-105-6]](/bjoc/content/figures/1860-5397-20-105-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Molecular structure of 12. Thermal ellipsoids drawn at 50% probability. Selected bond angles (°): C(5)–Se(1)–C(5) 100.11 (14) , C(7)–O(1)–C(8) 115.4 (2), O(2)–C(7)–O(1) 121.7 (2), O(2)–C(7)–C(1) 124.7 (2), N(1)–C(2)–C(1) 123.1 (2), N(1)–C(2)–C(3) 119.3 (2).
Figure 6: Molecular structure of 12. Thermal ellipsoids drawn at 50% probability. Selected bond angles (°): C...
Computational studies
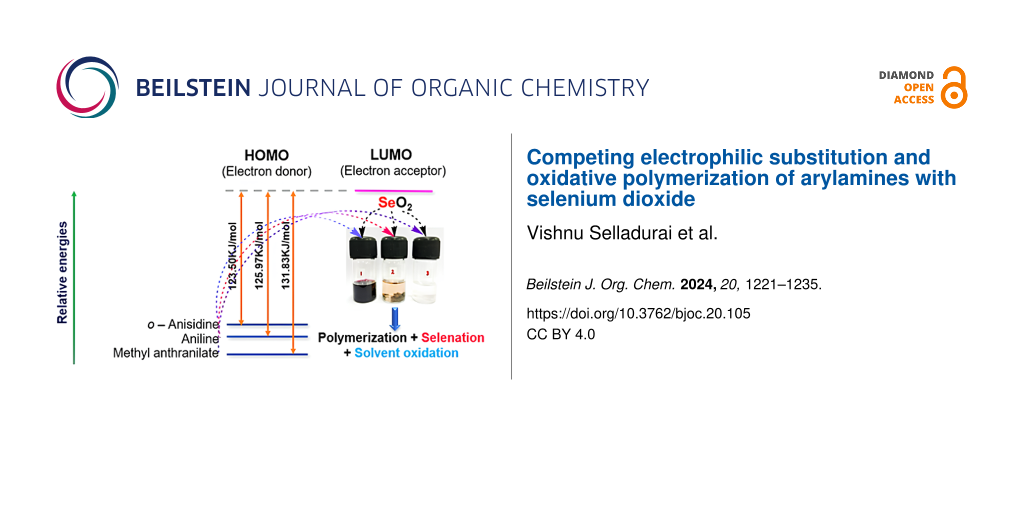
To comprehend the role of electron-donating and -withdrawing groups in arylamine oxidation using SeO2 as oxidant, DFT calculations were carried out on the arylamines and SeO2 using Gaussian 16 [60]. The molecular structure of aniline, o-anisidine, and methyl anthranilate were optimized using the B3LYP/6-31G(d,p) basis set. Oxidative electron transfer from an arylamine to SeO2 was the first step in the oxidation process. This step was especially crucial for the polymerization of arylamines. Hence, we proposed that comparing the relative energy of the frontier molecular orbitals of arylamines with SeO2 could be a convenient measure to understand the differences in polymerization tendency. The relative energy of the frontier molecular orbitals of arylamines and SeO2 is shown in Figure 7. In an oxidative polymerization process, an electron is transferred from the HOMO of an arylamine donor to the LUMO of the SeO2 acceptor. The computed HOMO–LUMO energy difference (∆E) between arylamines and SeO2 decreased in the following order: o-anisidine − SeO2 > aniline − SeO2 > methyl anthranilate − SeO2. It revealed that the HOMO of methyl anthranilate was relatively buried, with a large energy gap (mismatch) between the donor and acceptor orbitals, which slowed down the oxidative polymerization process and allowed the alternative reaction pathway, electrophilic substitution of SeO2 on the aryl ring. This result was further supported by our observation (vide supra) that the polymerization of methyl anthranilate was extremely slow and yielded a lower quantity of polymer after 24 h reaction time.
![[1860-5397-20-105-7]](/bjoc/content/figures/1860-5397-20-105-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Relative energy levels of arylamines and SeO2.
Figure 7: Relative energy levels of arylamines and SeO2.
In the next step, natural bond orbital (NBO) analysis was carried out to understand the effect of electron delocalization on the arylamine reactivity. The NBO analysis was carried out using the same basis set, B3LYP/6-31G(d,p). The natural charge (q) of the nitrogen atom, occupancy of the nitrogen lone pair orbital, second-order perturbation energy (E) for intramolecular donor–acceptor interactions, and the donation of electron density form Lewis orbitals to non-Lewis orbitals obtained via NBO analysis of arylamines are provided in Table 1 [61]. The partial negative charge of the nitrogen atom was the lowest for methyl anthranilate when compared to aniline and o-anisidine. It was further correlated to the relatively low lone pair occupancy of methyl anthranilate, indicating poor electron density availability of the nitrogen atom for oxidation by SeO2. Therefore, the SeO2-mediated polymerization was slow for methyl anthranilate. The poor electron density of the nitrogen atom was due to the significant deviation of the methyl anthranilate structure from the ideal Lewis structure. The lone pair electron of nitrogen was donated to the non-Lewis orbital (i.e., LP*) of the adjacent carbon C(4) (see Figure 8), with a high interaction energy of 93.62 kcal⋅mol−1. Such an interaction was not found for aniline and o-anisidine. Instead, for o-anisidine and aniline, the lone pair electrons were delocalized into the antibonding NBO (i.e., BD*) with a smaller interaction energy of ≈26 kcal⋅mol−1. In other words, as shown in Scheme 8, the lone electron pair present at the nitrogen atom was delocalized into the π-conjugated system.
Table 1: Summary of NBO analysis.
| arylamine | natural charge (q) of N | occupancy of N lone pair orbital | donor NBO | acceptor NBO | perturbation energy (E), kcal⋅mol−1 |
| aniline | −0.83507 | 1.85302 | LP–N12 | BD*–C3–C4 | 25.99 |
| o-anisidine | −0.84127 | 1.85122 | LP–N16 | BD*–C5–C6 | 25.64 |
| methyl anthranilate | −0.81177 | 1.75677 | LP–N11 | LP*–C4 | 93.62 |
![[1860-5397-20-105-8]](/bjoc/content/figures/1860-5397-20-105-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Computationally optimized structure of aniline (a), o-anisidine (b), and methyl anthranilate (c), with atom labels indicated.
Figure 8: Computationally optimized structure of aniline (a), o-anisidine (b), and methyl anthranilate (c), w...
![[1860-5397-20-105-i8]](/bjoc/content/inline/1860-5397-20-105-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Resonance structures for the delocalization of the nitrogen lone pair into the π-system.
Scheme 8: Resonance structures for the delocalization of the nitrogen lone pair into the π-system.
Conclusion
Aniline and o-anisidine underwent a predominant oxidative polymerization reaction with SeO2. Therein, electrophilic selenation was poor. The reactivity of the NH2 unit towards oxidation could be partially suppressed by the presence of an electron-withdrawing function. Consequently, for methyl anthranilate, polymerization was suppressed, and selenated compounds were obtained with an appreciable yield. Further, when SeO2 was used as electrophile, solvent oxidation (e.g., interaction between solvent and acidic α-CH unit) and oxidation of reactive functions (e.g., NH2 group) had to be taken into account. Characterization of the organoselenium compounds by HRMS, 1H, and 13C NMR was supported by 77Se NMR and single-crystal X-ray analysis in order to confirm the identity of the compounds.
Experimental
General procedures
All syntheses were carried out using the standard Schlenk line in a nitrogen environment. Acetonitrile (99.9%) was bought from Avra Chemicals Private Ltd. and used without any further purification. Selenium dioxide and a range of reactants were purchased from Sigma-Aldrich. TLC was performed using silica-gel-coated aluminum sheets (TLC silica gel 60 F254). 1H, 13C, and 77Se NMR (500, 126, and 96 MHz) spectra were recorded in CDCl3 on a Bruker Avance III 500 MHz device. The chemical shift δ is reported in ppm, and the multiplicity of the signals is abbreviated as singlet (s), doublet (d), double of doublets (dd), triplet of doublets (td), etc. The coupling constants J are expressed in Hz. The calibration was done with respect to the signal of residual undeuterated solvent or in relationship to TMS (CDCl3: 1H 7.26 ppm, 13C 77.34 ppm). Single-crystal X–ray diffraction was recorded on a Bruker D8 Quest system. HRESIMS spectra were recorded on a Waters Xevo G2-XS QTof device. UV–vis spectra of all polymers were recorded on a PerkinElmer Lambda 365 UV–vis spectrophotometer. FTIR spectra were recorded on an Agilent Cary 630 KBr module.
Reaction of aniline with SeO2
To a reaction vessel, 10 mL of acetonitrile were introduced along with aniline (10 mmol, ≈0.91 mL) and selenium dioxide (15 mmol, 1.66 g). The reaction mixture was stirred under a nitrogen atmosphere at 80 °C for 3 h. Before the solvent was evaporated at reduced pressure using a rotary evaporator, TLC was performed to verify completion of the reaction using 7:3 petroleum ether/ethyl acetate as the eluent. Upon evaporation of solvent, polymer 1 was obtained as a black solid. The solid was vacuum-filtered using a Büchner funnel and washed with acetonitrile. The solid was air-dried to give 2.33 g of a black crystalline solid that was insoluble in ethyl acetate and barely soluble in methanol. The filtrate was collected in a different round-bottom flask, and the solvent was evaporated in a rotary evaporator. The resulting solid was dissolved in DCM, and silica gel was added to the DCM layer. This slurry was subjected to column chromatography on silica gel (100–200 mesh) using petroleum ether and ethyl acetate as eluent. The initial pale yellow fraction was collected with 16% ethyl acetate, and the solvent was evaporated to give an off-white solid. Upon recrystallization from ethyl acetate, the above fraction afforded N1,N2-diphenyloxalamide (3) as a colorless solid. The remaining fractions were collected with 22% ethyl acetate. TLC suggested those fractions to be hardly separable. All fractions obtained with 20% ethyl acetate were combined and evaporated to give a black solid that contained a mixture of compounds, including the monoselenides 1 and 2.
Mixture of 4,4'-selenodianiline (1) and 2-((4-aminophenyl)selanyl)aniline (2): Black solid (28.2 mg); 77Se NMR (95 MHz, DMSO-d6, δ) 371.2, 296.1; HRESIMS (m/z): [M + H]+ calcd for C12H13N2Se (ortho/para), 265.0238; found, 265.0229, [M + H − PhNH2]+ calcd for C6H6NSe (ortho/para), 172.0238; found, 171.9667; FTIR (KBr) ν̃max: 3436, 3354, 3220, 3026, 1595, 1490, 1282, 1073, 820, 752 cm−1.
N1,N2-Diphenyloxalamide (3): Colorless solid (2.6 mg, 0.22%); mp 231–232 °C; 1H NMR (500 MHz, CDCl3, δ) 9.36 (s, 2H), 7.70 (d, J = 7.9 Hz, 4H), 7.44 (t, J = 7.8 Hz, 4H), 7.25 (t, J = 7.5 Hz, 2H); HRESIMS (m/z): [M + CH3COOH + H]+ calcd for C16H17N2O4, 301.1183; found, 301.1408, [2M + K]+ calcd for C28H24KN4O4, 519.1429; found, 518.8590; FTIR (KBr) ν̃max: 3354, 3063, 3019, 2840, 1684, 1602, 1528, 1252, 1021, 760 cm−1.
Reaction of o-anisidine with SeO2
A round-bottom flask was charged with 10 mL of acetonitrile, o-anisidine (10 mmol, ≈1.13 mL), and selenium dioxide (15 mmol, ≈1.66 g). The flask content was stirred under a nitrogen atmosphere at 80 °C for 3 h. Evaporation of the reaction mixture on a rotary evaporator gave a black crystalline solid. The resulting solid was vacuum-filtered, washed with acetonitrile, and air-dried to yield 2.36 g of polymer 2 as a black solid. The filtrate was evaporated and column-chromatographed on silica gel (100–200 mesh) using petroleum ether and ethyl acetate as an eluent. The initial yellowish orange fraction containing N1,N2-bis(2-methoxyphenyl)oxalamide, collected with 22% ethyl acetate, was recrystallized from a chloroform/methanol mixture. The second reddish brown fraction containing 2,5-bis((2-methoxyphenyl)amino)cyclohexa-2,5-diene-1,4-dione was collected with 24% ethyl acetate. After evaporation of the solvent, the reddish brown precipitate was recrystallized from chloroform/methanol and ethyl acetate mixture as solvent. The fractions obtained with 32% ethyl acetate were mixtures of compounds. These fractions were combined and evaporated to afford a black semisolid containing 4–8.
Mixture of monoselenides 4 and 5, diselenides 6 and 7, and selenoxide 8: Black solid (32 mg); 77Se NMR (95 MHz, CDCl3, δ) 792.52, 567.62, 537.42, 422.36, 405.91; HRESIMS (m/z): [M + H]+ calcd for C14H17N2O2Se (from 4 and 5), 325.0450; found, 325.0497, [M + H]+ calcd for C14H17N2O3Se (from 8), 341.0399; found, 341.0399, [M + H − PhNH2OMe]+ calcd for C7H8NOSe (from 4 and 5), 201.9800; found, 201.1051; FTIR (KBr) ν̃max: 3436, 2922, 2855, 1617, 1580, 1498, 1222, 1028, 812, 745 cm−1.
N1,N2-Bis(2-methoxyphenyl)oxalamide (9): Yellow solid (4.2 mg, 0.29%); mp 220–221 °C; 1H NMR (500 MHz, CDCl3, δ) 9.96 (s, 2H), 8.42 (dd, J = 8.1, 1.7 Hz, 2H), 7.14 (td, J = 7.8, 1.6 Hz, 2H), 7.02 (td, J = 7.8, 1.4 Hz, 2H), 6.94 (dd, J = 8.2, 1.4 Hz, 2H), 3.94 (s, 6H); 13C NMR (126 MHz, CDCl3, δ) 157.5, 148.9, 126.2, 125.3, 121.0, 119.7, 110.3, 55.8; HRESIMS (m/z): [M + Na]+ calcd for C16H16N2NaO4, 323.1002; found, 323.1006, [2M + Na]+ calcd for C32H32N4NaO8, 623.2112; found, 623.2112, [3M + Na]+ calcd for C48H48N6NaO12, 923.3222; found, 923.3201; FTIR (KBr) ν̃max: 3354, 1684, 1602, 1528, 1461, 1252, 1021, 760 cm−1.
2,5-Bis((2-methoxyphenyl)amino)cyclohexa-2,5-diene-1,4-dione (10): Reddish brown solid (49 mg, 2.8%); mp 237–239 °C; 1H NMR (500 MHz, CDCl3, δ) 8.48 (s, 2H), 7.41 (dd, J = 7.9, 1.5 Hz, 2H), 7.16 (td, J = 7.9, 1.5 Hz, 2H), 7.04–6.93 (m, 4H), 6.17 (s, 2H), 3.91 (s, 6H); 13C NMR (126 MHz, CDCl3, δ) 180.6, 151.4, 145.6, 126.7, 125.9, 121.2, 120.8, 111.2, 96.4, 55.8; HRESIMS (m/z): [M + H]+ calcd for C20H19N2O4, 351.1339; found, 351.1350; FTIR (KBr) ν̃max: 3429, 3324, 3026, 2937, 1647, 1580, 1252, 1028, 738 cm−1.
Reaction of methyl anthranilate with SeO2
To a round-bottom flask, 10 mL of acetonitrile were introduced along with methyl anthranilate (10 mmol, ≈1.29 mL) and selenium dioxide (15 mmol, ≈1.66 g). The reaction mixture was stirred under a nitrogen atmosphere at 80 °C for 24 h. The reaction progress was monitored using TLC. Evaporation of the reaction mixture on a rotary evaporator afforded polymer 3 as a semisolid along with other compounds. The semisolid was triturated with a 2:8 mixture of ethyl acetate and petroleum ether as solvent. Then, the supernatant was decanted into a beaker. The semisolid was allowed to dry, giving polymer 3 as black solid (0.36 g). The supernatant was concentrated, redissolved in DCM, and turned into a slurry by addition of silica gel (100–200 mesh) The slurry was subjected to column chromatography using petroleum ether and ethyl acetate as eluent. The first minor fraction was collected with 18% ethyl acetate. Upon evaporation of the solvent, dimethyl 2,2'-(oxalylbis(azanediyl))dibenzoate was obtained as a pale yellow solid and recrystallized from a 9:1 methanol/chloroform mixture. The second major fraction was collected with 22% ethyl acetate. Evaporation of the solvent afforded methyl 2-amino-3-((4-amino-3-(methoxycarbonyl)phenyl)selanyl)benzoate as pale yellow solid. It was recrystallized from a 9:1 methanol/chloroform mixture. The third major fraction was also collected with 22% ethyl acetate. After evaporation of the solvent, dimethyl 5,5'-selenobis(2-aminobenzoate) was obtained as yellow solid. The solid was recrystallized from a 9:1 methanol/chloroform mixture to afford a pale yellow solid.
Methyl 2-amino-3-((4-amino-3-(methoxycarbonyl)phenyl)selanyl)benzoate (11): Pale yellow solid (87 mg, 4.6%); mp 152–153 °C; 1H NMR (500 MHz, CDCl3, δ) 8.02 (d, J = 2.2 Hz, 1H), 7.88 (dd, J = 8.1, 1.7 Hz, 1H), 7.68 (dd, J = 7.5, 1.7 Hz, 1H), 7.24 (dd, J=8.6, J = 2.1 Hz, 1H), 6.59–6.51 (m, 2H), 3.85 (s, 3H), 3.84 (s, 3H); 13C NMR (126 MHz, CDCl3) 168.5, 167.9, 150.9, 150.0, 142.2, 138.2, 135.5, 132.6, 118.0, 117.3, 116.0, 114.7, 111.4, 110.8, 51.7; 77Se NMR (95 MHz, CDCl3, δ) 300.2. HRESIMS (m/z): [M + H]+ calcd for C16H17N2O4Se, 381.0348; found, 381.0359, [M + H − PhNH2CO2Me]+ calcd for C8H8NO2Se, 229.9715; found, 229.9721; FTIR (KBr) ν̃max: 3213, 3078, 1699, 1580, 1505, 1267, 820 cm−1.
Dimethyl 5,5'-selenobis(2-aminobenzoate) (12): Yellow solid (476 mg, 25.1%); mp 163–164 °C; 1H NMR (500 MHz, CDCl3, δ) 8.06 (d, J = 2.1 Hz, 2H), 7.35 (dd, J = 8.6, 2.1 Hz, 2H), 6.55 (d, J = 8.5 Hz, 2H), 5.77 (s, 4H), 3.84 (s, 6H); 13C NMR (126 MHz, CDCl3, δ) 168.0, 145.0, 139.5, 136.5, 117.8, 116.8, 111.3, 51.7; 77Se NMR (95 MHz, CDCl3, δ) 394.1; HRESIMS (m/z): [M + H]+ calcd for C16H17N2O4Se, 381.0348; found, 381.0356; FTIR (KBr) ν̃max: 3466, 3377, 1677, 1617, 1413, 1237, 1095, 752 cm−1.
Dimethyl 2,2'-(oxalylbis(azanediyl))dibenzoate (13): Pale yellow solid (1.9 mg, 0.12%); mp 251–252 °C; 1H NMR (500 MHz, CDCl3, δ) 12.89 (s, 2H), 8.88 (dd, J = 8.4, 1.2 Hz, 2H), 8.11 (dd, J = 7.9, 1.6 Hz, 2H), 7.61 (ddd, J = 8.7, 7.3, 1.7 Hz, 2H), 7.24–7.17 (m, 2H), 4.01 (s, 6H); 13C NMR (126 MHz, CDCl3, δ) 168.1, 158.4, 139.6, 134.6, 131.2, 124.0, 120.5, 116.6, 52.7; HRESIMS (m/z): [M + Na]+ calcd for C18H16N2NaO6, 379.0901; found, 379.0924, [2M + Na]+ calcd for C36H32N4NaO12, 735.1909; found, 735.1898, [3M + Na]+ calcd for C54H48N6NaO18, 1091.2917; found, 1091.2885; FTIR (KBr) ν̃max: 3444, 3354, 1677, 1625, 1423, 1304, 1237, 820 cm−1.
X-ray data
Table 2 and Table 3 show single-crystal X-ray structure refinement data.
Table 2: Single-crystal X-ray structure refinement data for 3, 9, and 10.
| parameter | 3 | 9 | 10 |
| empirical formula | C14H12N2O2 | C16H16N2O4 | C20H18N2O4 |
| formula weight | 240.26 | 300.31 | 350.36 |
| crystal system | monoclinic | monoclinic | monoclinic |
| space group | P21/n | P21/c | C2/c |
| a, Å | 5.3310(9) | 7.7817(10) | 19.843(4) |
| b, Å | 5.3762(9) | 14.954(2) | 3.8540(8) |
| c, Å | 20.182(3) | 6.9263(9) | 21.797(5) |
| α, ° | 90 | 90 | 90 |
| β, ° | 93.598(5) | 114.369(4) | 96.420(8) |
| γ, ° | 90 | 90 | 90 |
| V, Å3 | 577.29(17) | 734.19(17) | 1656.5 (6) |
| Z | 2 | 2 | 4 |
| ρ calcd, mg⋅m−3 | 1.382 | 1.358 | 1.405 |
| absorption coefficient, mm−1 | 0.094 | 0.099 | 0.099 |
| reflections collected | 14639 | 16216 | 16841 |
| final R(F) [I > 2σ(I)] | 0.0570 | 0.0866 | 0.0615 |
| wR(F2) indices [I > 2σ(I)] | 0.1209 | 0.1303 | 0.1249 |
| data/restraints/parameters | 1410/0/82 | 1838/0/101 | 2061/0/122 |
| goodness-of-fit on F2 | 1.072 | 1.051 | 1.068 |
Table 3: Single-crystal X-ray structure refinement data for 11, 12, and 13.
| parameter | 11 | 12 | 13 |
| empirical formula | C16H16N2O4Se | C16H16N2O4Se | C18H16N2O6 |
| formula weight | 379.27 | 379.27 | 356.33 |
| crystal system | triclinic | orthorhombic | triclinic |
| space group | P−1 | P21212 | P−1 |
| a, Å | 5.2732(10) | 9.2080(17) | 5.4243(6) |
| b, Å | 11.353(2) | 19.174(3) | 8.3905(10) |
| c, Å | 14.331(3) | 4.5051(7) | 9.2803(11) |
| α, ° | 105.300(6) | 90 | 84.202(4) |
| β, ° | 97.375(6) | 90 | 84.144(4) |
| γ, ° | 101.622(6) | 90 | 75.208(4) |
| V, Å3 | 795.5(3) | 795.4 (2) | 405.01(8) |
| Z | 2 | 2 | 1 |
| ρ calcd, mg⋅m−3 | 1.583 | 1.584 | 1.461 |
| absorption coefficient, mm−1 | 2.382 | 2.382 | 0.111 |
| reflections collected | 20734 | 31206 | 14437 |
| final R(F) [I > 2σ(I)] | 0.0352 | 0.0246 | 0.0438 |
| wR(F2) indices [I > 2σ(I)] | 0.0726 | 0.0586 | 0.1029 |
| data/restraints/parameters | 3595/0/210 | 1638/0/106 | 1646/0/119 |
| goodness-of-fit on F2 | 1.035 | 1.095 | 1.039 |
Supporting Information
| Supporting Information File 1: Spectroscopic characterization of products (1H, 13C and 77Se NMR, IR, and HRMS spectra), packing arrangements of compounds and HOMO and LUMO energy values for reactants. | ||
| Format: PDF | Size: 2.8 MB | Download |
| Supporting Information File 2: Crystallographic data for compound 3. | ||
| Format: CIF | Size: 475.1 KB | Download |
| Supporting Information File 3: Crystallographic data for compound 9. | ||
| Format: CIF | Size: 505.0 KB | Download |
| Supporting Information File 4: Crystallographic data for compound 10. | ||
| Format: CIF | Size: 558.6 KB | Download |
| Supporting Information File 5: Crystallographic data for compound 11. | ||
| Format: CIF | Size: 627.2 KB | Download |
| Supporting Information File 6: Crystallographic data for compound 12. | ||
| Format: CIF | Size: 937.3 KB | Download |
| Supporting Information File 7: Crystallographic data for compound 13. | ||
| Format: CIF | Size: 439.8 KB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Sies, H. Redox Biol. 2015, 4, 180–183. doi:10.1016/j.redox.2015.01.002
Return to citation in text: [1] -
Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2180. doi:10.1021/cr000426w
Return to citation in text: [1] -
Alexander, V.; Choi, W. J.; Chun, J.; Kim, H. O.; Jeon, J. H.; Tosh, D. K.; Lee, H. W.; Chandra, G.; Choi, J.; Jeong, L. S. Org. Lett. 2010, 12, 2242–2245. doi:10.1021/ol1005906
Return to citation in text: [1] -
Thun-Hohenstein, S. T. D.; Suits, T. F.; Malla, T. R.; Tumber, A.; Brewitz, L.; Choudhry, H.; Salah, E.; Schofield, C. J. ChemMedChem 2022, 17, e202100582. doi:10.1002/cmdc.202100582
Return to citation in text: [1] -
Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. J. Med. Chem. 2017, 60, 7300–7314. doi:10.1021/acs.jmedchem.7b00480
Return to citation in text: [1] -
Banerjee, K.; Padmavathi, G.; Bhattacherjee, D.; Saha, S.; Kunnumakkara, A. B.; Bhabak, K. P. Org. Biomol. Chem. 2018, 16, 8769–8782. doi:10.1039/c8ob01891j
Return to citation in text: [1] -
Iwaoka, M.; Tomoda, S. J. Am. Chem. Soc. 1994, 116, 2557–2561. doi:10.1021/ja00085a040
Return to citation in text: [1] -
Doig, A. I.; Tuck, T. A.; LeBlanc, B.; Back, T. G. ACS Omega 2022, 7, 27312–27323. doi:10.1021/acsomega.2c02286
Return to citation in text: [1] -
Singh, V. P.; Poon, J.-f.; Yan, J.; Lu, X.; Ott, M. K.; Butcher, R. J.; Gates, P. J.; Engman, L. J. Org. Chem. 2017, 82, 313–321. doi:10.1021/acs.joc.6b02418
Return to citation in text: [1] -
Sands, K. N.; Tuck, T. A.; Back, T. G. Chem. – Eur. J. 2018, 24, 9714–9728. doi:10.1002/chem.201800182
Return to citation in text: [1] -
Yoshida, S.; Kumakura, F.; Komatsu, I.; Arai, K.; Onuma, Y.; Hojo, H.; Singh, B. G.; Priyadarsini, K. I.; Iwaoka, M. Angew. Chem. 2011, 123, 2173–2176. doi:10.1002/ange.201006939
Return to citation in text: [1] -
Iwaoka, M.; Shimodaira, S. Synthesis and catalytic functions of selenopeptides. In Organochalcogen Compounds; Lenardão, E. J.; Perin, G.; Santi, C.; Alves, D., Eds.; Elsevier: Amsterdam, Netherlands, 2022; pp 195–218. doi:10.1016/b978-0-12-819449-2.00004-5
Return to citation in text: [1] -
da Silva, R. B.; Lange Coelho, F.; Rodembusch, F. S.; Schwab, R. S.; Schneider, J. M. F. M.; da Silveira Rampon, D.; Schneider, P. H. New J. Chem. 2019, 43, 11596–11603. doi:10.1039/c9nj01948k
Return to citation in text: [1] -
Mugesh, G.; Singh, H. B. Acc. Chem. Res. 2002, 35, 226–236. doi:10.1021/ar010091k
Return to citation in text: [1] -
Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Molecules 2019, 24, 4484. doi:10.3390/molecules24244484
Return to citation in text: [1] -
Balkrishna, S. J.; Bhakuni, B. S.; Chopra, D.; Kumar, S. Org. Lett. 2010, 12, 5394–5397. doi:10.1021/ol102027j
Return to citation in text: [1] -
Wang, Y.; Deng, J.; Chen, J.; Cao, F.; Hou, Y.; Yang, Y.; Deng, X.; Yang, J.; Wu, L.; Shao, X.; Shi, T.; Wang, Z. ACS Catal. 2020, 10, 2707–2712. doi:10.1021/acscatal.9b04931
Return to citation in text: [1] -
Matsumura, M.; Takahashi, T.; Yamauchi, H.; Sakuma, S.; Hayashi, Y.; Hyodo, T.; Obata, T.; Yamaguchi, K.; Fujiwara, Y.; Yasuike, S. Beilstein J. Org. Chem. 2020, 16, 1075–1083. doi:10.3762/bjoc.16.94
Return to citation in text: [1] -
Selvakumar, K.; Singh, V. P.; Shah, P.; Singh, H. B. Main Group Chem. 2011, 10, 141–152. doi:10.3233/mgc-2011-044
Return to citation in text: [1] -
Singh, V. P.; Poon, J.-f.; Butcher, R. J.; Lu, X.; Mestres, G.; Ott, M. K.; Engman, L. J. Org. Chem. 2015, 80, 7385–7395. doi:10.1021/acs.joc.5b00797
Return to citation in text: [1] -
Duarte, L. F. B.; Oliveira, R. L.; Rodrigues, K. C.; Voss, G. T.; Godoi, B.; Schumacher, R. F.; Perin, G.; Wilhelm, E. A.; Luchese, C.; Alves, D. Bioorg. Med. Chem. 2017, 25, 6718–6723. doi:10.1016/j.bmc.2017.11.019
Return to citation in text: [1] -
Sakthinathan, I.; Mahendran, M.; Krishnan, K.; Karuthapandi, S. Sustainable Energy Fuels 2021, 5, 3617–3631. doi:10.1039/d1se00595b
Return to citation in text: [1] -
Freudendahl, D. M.; Santoro, S.; Shahzad, S. A.; Santi, C.; Wirth, T. Angew. Chem., Int. Ed. 2009, 48, 8409–8411. doi:10.1002/anie.200903893
Return to citation in text: [1] -
Wirth, T., Ed. Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527641949
Return to citation in text: [1] -
Santi, C.; Scimmi, C. Selenium and Tellurium Complexes in Organic Synthesis. In Comprehensive Organometallic Chemistry IV; Parkin, G.; Meyer, K.; O’Hare, D., Eds.; Elsevier: Amsterdam, Netherlands, 2022; pp 536–562. doi:10.1016/b978-0-12-820206-7.00082-2
Return to citation in text: [1] -
Belladona, A. L.; Cervo, R.; Alves, D.; Barcellos, T.; Cargnelutti, R.; Schumacher, R. F. Tetrahedron Lett. 2020, 61, 152035. doi:10.1016/j.tetlet.2020.152035
Return to citation in text: [1] -
Kour, J.; Khajuria, P.; Verma, P. K.; Kapoor, N.; Kumar, A.; Sawant, S. D. ACS Omega 2022, 7, 13000–13009. doi:10.1021/acsomega.2c00323
Return to citation in text: [1] [2] -
Wanniang, K.; Shangpliang, O. R.; Marpna, I. D.; Lipon, T. M.; Laloo, B. M.; Myrboh, B. J. Org. Chem. 2020, 85, 15652–15659. doi:10.1021/acs.joc.0c02028
Return to citation in text: [1] [2] -
Ali, D.; Mondal, N.; Panday, A. K.; Choudhury, L. H. ACS Omega 2023, 8, 25349–25357. doi:10.1021/acsomega.3c02769
Return to citation in text: [1] -
Thurow, S.; Penteado, F.; Perin, G.; Alves, D.; Santi, C.; Monti, B.; Schiesser, C. H.; Barcellos, T.; Lenardão, E. J. Org. Chem. Front. 2018, 5, 1983–1991. doi:10.1039/c8qo00360b
Return to citation in text: [1] -
Dibrell, S. E.; Maser, M. R.; Reisman, S. E. J. Am. Chem. Soc. 2020, 142, 6483–6487. doi:10.1021/jacs.9b13818
Return to citation in text: [1] -
Pathe, G. K.; Ahmed, N. RSC Adv. 2015, 5, 59114–59119. doi:10.1039/c5ra09986b
Return to citation in text: [1] -
Ren, Y.; Xu, B.; Zhong, Z.; Pittman, C. U., Jr.; Zhou, A. Org. Chem. Front. 2019, 6, 2023–2027. doi:10.1039/c9qo00299e
Return to citation in text: [1] -
Kumar, R. U.; Reddy, K. H. V.; Satish, G.; Swapna, K.; Nageswar, Y. V. D. Tetrahedron Lett. 2016, 57, 4138–4141. doi:10.1016/j.tetlet.2016.07.075
Return to citation in text: [1] -
Hu, M.; Ren, Y. Org. Biomol. Chem. 2021, 19, 6692–6696. doi:10.1039/d1ob01070k
Return to citation in text: [1] -
Quell, T.; Mirion, M.; Schollmeyer, D.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. ChemistryOpen 2016, 5, 115–119. doi:10.1002/open.201500206
Return to citation in text: [1] -
Bhat, M. Y.; Kumar, A.; Ahmed, Q. N. Tetrahedron 2020, 76, 131105. doi:10.1016/j.tet.2020.131105
Return to citation in text: [1] [2] [3] -
Kim, H.; Ku, B.-C.; Goh, M.; Ko, H. C.; Ando, S.; You, N.-H. Macromolecules 2019, 52, 827–834. doi:10.1021/acs.macromol.8b02139
Return to citation in text: [1] -
Selvakumar, K.; Singh, H. B.; Goel, N.; Singh, U. P.; Butcher, R. J. Dalton Trans. 2011, 40, 9858–9867. doi:10.1039/c1dt10862j
Return to citation in text: [1] [2] [3] -
Selvakumar, K.; Shah, P.; Singh, H. B.; Butcher, R. J. Chem. – Eur. J. 2011, 17, 12741–12755. doi:10.1002/chem.201100930
Return to citation in text: [1] -
Ding, Y.; Liang, J.; Liu, G.; Ni, W.; Shen, L. Coatings 2019, 9, 399. doi:10.3390/coatings9060399
Return to citation in text: [1] -
Baker, A. G. ARO (Koya) 2019, 7, 47–52. doi:10.14500/aro.10483
Return to citation in text: [1] -
Dominic, J.; David, T.; Vanaja, A.; Satheesh Kumar, K. K. Eur. Polym. J. 2016, 85, 236–243. doi:10.1016/j.eurpolymj.2016.10.028
Return to citation in text: [1] -
Ibrahim, K. A. Arabian J. Chem. 2017, 10, S2668–S2674. doi:10.1016/j.arabjc.2013.10.010
Return to citation in text: [1] -
Manimegalai, P.; Dominic, J.; Satheesh Kumar, K. K. Polym. Int. 2022, 71, 770–776.
Return to citation in text: [1] -
Tanini, D.; Dalia, C.; Capperucci, A. Green Chem. 2021, 23, 5680–5686. doi:10.1039/d0gc04322b
Return to citation in text: [1] -
Li, Q.; Zhang, J.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Polymers (Basel, Switz.) 2018, 10, 417. doi:10.3390/polym10040417
Return to citation in text: [1] -
Won, M.-S.; Yeom, J.-S.; Yoon, J.-H.; Jeong, E.-D.; Shim, Y.-B. Bull. Korean Chem. Soc. 2003, 24, 948–952. doi:10.5012/bkcs.2003.24.7.948
Return to citation in text: [1] -
Wen, Y.-H.; Li, X.-M.; Wang, L.; Zhang, S.-S. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o2185–o2186. doi:10.1107/s1600536806015972
Return to citation in text: [1] [2] -
Santana, M. D.; García, G.; Vicente-Hernández, C.; García, L.; Pérez, J.; Rojo, T.; Lezama, L. J. Organomet. Chem. 2008, 693, 2009–2016. doi:10.1016/j.jorganchem.2008.03.001
Return to citation in text: [1] -
Zhou, W.; Fan, M.; Yin, J.; Jiang, Y.; Ma, D. J. Am. Chem. Soc. 2015, 137, 11942–11945. doi:10.1021/jacs.5b08411
Return to citation in text: [1] -
González-Ibarra, M.; Farfán, N.; Trejo, C.; Uribe, S.; Lotina-Hennsen, B. J. Agric. Food Chem. 2005, 53, 3415–3420. doi:10.1021/jf047883o
Return to citation in text: [1] -
Rodrigues, N. D. N.; Cole-Filipiak, N. C.; Horbury, M. D.; Staniforth, M.; Karsili, T. N. V.; Peperstraete, Y.; Stavros, V. G. J. Photochem. Photobiol., A 2018, 353, 376–384. doi:10.1016/j.jphotochem.2017.11.042
Return to citation in text: [1] -
Black, D. S. C.; Vanderzalm, C. H. B.; Hartshorn, A. J. Aust. J. Chem. 1977, 30, 225–229. doi:10.1071/ch9770225
Return to citation in text: [1] -
Orsy, G.; Fülöp, F.; Mándity, I. M. Molecules 2020, 25, 1985. doi:10.3390/molecules25081985
Return to citation in text: [1] -
Pujato, N.; Díaz, G.; Barbagelata, M. S.; Vicco, M. H.; Calvinho, L. F.; Marcipar, I. S. Appl. Biochem. Biotechnol. 2015, 175, 141–154. doi:10.1007/s12010-014-1246-y
Return to citation in text: [1] -
Panis, F.; Kampatsikas, I.; Bijelic, A.; Rompel, A. Sci. Rep. 2020, 10, 1659. doi:10.1038/s41598-020-57671-x
Return to citation in text: [1] -
Samuels, N. M.; Klinman, J. P. Biochemistry 2005, 44, 14308–14317. doi:10.1021/bi051176m
Return to citation in text: [1] -
Kumar, V.; Triglav, M.; Morin, V. M.; Bryce, D. L. ACS Org. Inorg. Au 2022, 2, 252–260. doi:10.1021/acsorginorgau.1c00051
Return to citation in text: [1] -
Gaussian 16, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2019.
Return to citation in text: [1] -
Fernández Riveras, J. A.; Frontera, A.; Bauzá, A. Phys. Chem. Chem. Phys. 2021, 23, 17656–17662. doi:10.1039/d1cp01929e
Return to citation in text: [1]
| 39. | Selvakumar, K.; Singh, H. B.; Goel, N.; Singh, U. P.; Butcher, R. J. Dalton Trans. 2011, 40, 9858–9867. doi:10.1039/c1dt10862j |
| 50. | Santana, M. D.; García, G.; Vicente-Hernández, C.; García, L.; Pérez, J.; Rojo, T.; Lezama, L. J. Organomet. Chem. 2008, 693, 2009–2016. doi:10.1016/j.jorganchem.2008.03.001 |
| 51. | Zhou, W.; Fan, M.; Yin, J.; Jiang, Y.; Ma, D. J. Am. Chem. Soc. 2015, 137, 11942–11945. doi:10.1021/jacs.5b08411 |
| 52. | González-Ibarra, M.; Farfán, N.; Trejo, C.; Uribe, S.; Lotina-Hennsen, B. J. Agric. Food Chem. 2005, 53, 3415–3420. doi:10.1021/jf047883o |
| 1. | Sies, H. Redox Biol. 2015, 4, 180–183. doi:10.1016/j.redox.2015.01.002 |
| 2. | Mugesh, G.; du Mont, W.-W.; Sies, H. Chem. Rev. 2001, 101, 2125–2180. doi:10.1021/cr000426w |
| 12. | Iwaoka, M.; Shimodaira, S. Synthesis and catalytic functions of selenopeptides. In Organochalcogen Compounds; Lenardão, E. J.; Perin, G.; Santi, C.; Alves, D., Eds.; Elsevier: Amsterdam, Netherlands, 2022; pp 195–218. doi:10.1016/b978-0-12-819449-2.00004-5 |
| 13. | da Silva, R. B.; Lange Coelho, F.; Rodembusch, F. S.; Schwab, R. S.; Schneider, J. M. F. M.; da Silveira Rampon, D.; Schneider, P. H. New J. Chem. 2019, 43, 11596–11603. doi:10.1039/c9nj01948k |
| 35. | Hu, M.; Ren, Y. Org. Biomol. Chem. 2021, 19, 6692–6696. doi:10.1039/d1ob01070k |
| 59. | Kumar, V.; Triglav, M.; Morin, V. M.; Bryce, D. L. ACS Org. Inorg. Au 2022, 2, 252–260. doi:10.1021/acsorginorgau.1c00051 |
| 7. | Iwaoka, M.; Tomoda, S. J. Am. Chem. Soc. 1994, 116, 2557–2561. doi:10.1021/ja00085a040 |
| 8. | Doig, A. I.; Tuck, T. A.; LeBlanc, B.; Back, T. G. ACS Omega 2022, 7, 27312–27323. doi:10.1021/acsomega.2c02286 |
| 9. | Singh, V. P.; Poon, J.-f.; Yan, J.; Lu, X.; Ott, M. K.; Butcher, R. J.; Gates, P. J.; Engman, L. J. Org. Chem. 2017, 82, 313–321. doi:10.1021/acs.joc.6b02418 |
| 10. | Sands, K. N.; Tuck, T. A.; Back, T. G. Chem. – Eur. J. 2018, 24, 9714–9728. doi:10.1002/chem.201800182 |
| 11. | Yoshida, S.; Kumakura, F.; Komatsu, I.; Arai, K.; Onuma, Y.; Hojo, H.; Singh, B. G.; Priyadarsini, K. I.; Iwaoka, M. Angew. Chem. 2011, 123, 2173–2176. doi:10.1002/ange.201006939 |
| 36. | Quell, T.; Mirion, M.; Schollmeyer, D.; Dyballa, K. M.; Franke, R.; Waldvogel, S. R. ChemistryOpen 2016, 5, 115–119. doi:10.1002/open.201500206 |
| 5. | Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. J. Med. Chem. 2017, 60, 7300–7314. doi:10.1021/acs.jmedchem.7b00480 |
| 6. | Banerjee, K.; Padmavathi, G.; Bhattacherjee, D.; Saha, S.; Kunnumakkara, A. B.; Bhabak, K. P. Org. Biomol. Chem. 2018, 16, 8769–8782. doi:10.1039/c8ob01891j |
| 33. | Ren, Y.; Xu, B.; Zhong, Z.; Pittman, C. U., Jr.; Zhou, A. Org. Chem. Front. 2019, 6, 2023–2027. doi:10.1039/c9qo00299e |
| 56. | Pujato, N.; Díaz, G.; Barbagelata, M. S.; Vicco, M. H.; Calvinho, L. F.; Marcipar, I. S. Appl. Biochem. Biotechnol. 2015, 175, 141–154. doi:10.1007/s12010-014-1246-y |
| 57. | Panis, F.; Kampatsikas, I.; Bijelic, A.; Rompel, A. Sci. Rep. 2020, 10, 1659. doi:10.1038/s41598-020-57671-x |
| 58. | Samuels, N. M.; Klinman, J. P. Biochemistry 2005, 44, 14308–14317. doi:10.1021/bi051176m |
| 3. | Alexander, V.; Choi, W. J.; Chun, J.; Kim, H. O.; Jeon, J. H.; Tosh, D. K.; Lee, H. W.; Chandra, G.; Choi, J.; Jeong, L. S. Org. Lett. 2010, 12, 2242–2245. doi:10.1021/ol1005906 |
| 4. | Thun-Hohenstein, S. T. D.; Suits, T. F.; Malla, T. R.; Tumber, A.; Brewitz, L.; Choudhry, H.; Salah, E.; Schofield, C. J. ChemMedChem 2022, 17, e202100582. doi:10.1002/cmdc.202100582 |
| 34. | Kumar, R. U.; Reddy, K. H. V.; Satish, G.; Swapna, K.; Nageswar, Y. V. D. Tetrahedron Lett. 2016, 57, 4138–4141. doi:10.1016/j.tetlet.2016.07.075 |
| 49. | Wen, Y.-H.; Li, X.-M.; Wang, L.; Zhang, S.-S. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o2185–o2186. doi:10.1107/s1600536806015972 |
| 23. | Freudendahl, D. M.; Santoro, S.; Shahzad, S. A.; Santi, C.; Wirth, T. Angew. Chem., Int. Ed. 2009, 48, 8409–8411. doi:10.1002/anie.200903893 |
| 24. | Wirth, T., Ed. Organoselenium Chemistry: Synthesis and Reactions; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527641949 |
| 25. | Santi, C.; Scimmi, C. Selenium and Tellurium Complexes in Organic Synthesis. In Comprehensive Organometallic Chemistry IV; Parkin, G.; Meyer, K.; O’Hare, D., Eds.; Elsevier: Amsterdam, Netherlands, 2022; pp 536–562. doi:10.1016/b978-0-12-820206-7.00082-2 |
| 27. | Kour, J.; Khajuria, P.; Verma, P. K.; Kapoor, N.; Kumar, A.; Sawant, S. D. ACS Omega 2022, 7, 13000–13009. doi:10.1021/acsomega.2c00323 |
| 28. | Wanniang, K.; Shangpliang, O. R.; Marpna, I. D.; Lipon, T. M.; Laloo, B. M.; Myrboh, B. J. Org. Chem. 2020, 85, 15652–15659. doi:10.1021/acs.joc.0c02028 |
| 29. | Ali, D.; Mondal, N.; Panday, A. K.; Choudhury, L. H. ACS Omega 2023, 8, 25349–25357. doi:10.1021/acsomega.3c02769 |
| 39. | Selvakumar, K.; Singh, H. B.; Goel, N.; Singh, U. P.; Butcher, R. J. Dalton Trans. 2011, 40, 9858–9867. doi:10.1039/c1dt10862j |
| 19. | Selvakumar, K.; Singh, V. P.; Shah, P.; Singh, H. B. Main Group Chem. 2011, 10, 141–152. doi:10.3233/mgc-2011-044 |
| 20. | Singh, V. P.; Poon, J.-f.; Butcher, R. J.; Lu, X.; Mestres, G.; Ott, M. K.; Engman, L. J. Org. Chem. 2015, 80, 7385–7395. doi:10.1021/acs.joc.5b00797 |
| 21. | Duarte, L. F. B.; Oliveira, R. L.; Rodrigues, K. C.; Voss, G. T.; Godoi, B.; Schumacher, R. F.; Perin, G.; Wilhelm, E. A.; Luchese, C.; Alves, D. Bioorg. Med. Chem. 2017, 25, 6718–6723. doi:10.1016/j.bmc.2017.11.019 |
| 22. | Sakthinathan, I.; Mahendran, M.; Krishnan, K.; Karuthapandi, S. Sustainable Energy Fuels 2021, 5, 3617–3631. doi:10.1039/d1se00595b |
| 30. | Thurow, S.; Penteado, F.; Perin, G.; Alves, D.; Santi, C.; Monti, B.; Schiesser, C. H.; Barcellos, T.; Lenardão, E. J. Org. Chem. Front. 2018, 5, 1983–1991. doi:10.1039/c8qo00360b |
| 31. | Dibrell, S. E.; Maser, M. R.; Reisman, S. E. J. Am. Chem. Soc. 2020, 142, 6483–6487. doi:10.1021/jacs.9b13818 |
| 32. | Pathe, G. K.; Ahmed, N. RSC Adv. 2015, 5, 59114–59119. doi:10.1039/c5ra09986b |
| 55. | Orsy, G.; Fülöp, F.; Mándity, I. M. Molecules 2020, 25, 1985. doi:10.3390/molecules25081985 |
| 16. | Balkrishna, S. J.; Bhakuni, B. S.; Chopra, D.; Kumar, S. Org. Lett. 2010, 12, 5394–5397. doi:10.1021/ol102027j |
| 17. | Wang, Y.; Deng, J.; Chen, J.; Cao, F.; Hou, Y.; Yang, Y.; Deng, X.; Yang, J.; Wu, L.; Shao, X.; Shi, T.; Wang, Z. ACS Catal. 2020, 10, 2707–2712. doi:10.1021/acscatal.9b04931 |
| 18. | Matsumura, M.; Takahashi, T.; Yamauchi, H.; Sakuma, S.; Hayashi, Y.; Hyodo, T.; Obata, T.; Yamaguchi, K.; Fujiwara, Y.; Yasuike, S. Beilstein J. Org. Chem. 2020, 16, 1075–1083. doi:10.3762/bjoc.16.94 |
| 53. | Rodrigues, N. D. N.; Cole-Filipiak, N. C.; Horbury, M. D.; Staniforth, M.; Karsili, T. N. V.; Peperstraete, Y.; Stavros, V. G. J. Photochem. Photobiol., A 2018, 353, 376–384. doi:10.1016/j.jphotochem.2017.11.042 |
| 14. | Mugesh, G.; Singh, H. B. Acc. Chem. Res. 2002, 35, 226–236. doi:10.1021/ar010091k |
| 15. | Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Molecules 2019, 24, 4484. doi:10.3390/molecules24244484 |
| 26. | Belladona, A. L.; Cervo, R.; Alves, D.; Barcellos, T.; Cargnelutti, R.; Schumacher, R. F. Tetrahedron Lett. 2020, 61, 152035. doi:10.1016/j.tetlet.2020.152035 |
| 27. | Kour, J.; Khajuria, P.; Verma, P. K.; Kapoor, N.; Kumar, A.; Sawant, S. D. ACS Omega 2022, 7, 13000–13009. doi:10.1021/acsomega.2c00323 |
| 28. | Wanniang, K.; Shangpliang, O. R.; Marpna, I. D.; Lipon, T. M.; Laloo, B. M.; Myrboh, B. J. Org. Chem. 2020, 85, 15652–15659. doi:10.1021/acs.joc.0c02028 |
| 54. | Black, D. S. C.; Vanderzalm, C. H. B.; Hartshorn, A. J. Aust. J. Chem. 1977, 30, 225–229. doi:10.1071/ch9770225 |
| 39. | Selvakumar, K.; Singh, H. B.; Goel, N.; Singh, U. P.; Butcher, R. J. Dalton Trans. 2011, 40, 9858–9867. doi:10.1039/c1dt10862j |
| 40. | Selvakumar, K.; Shah, P.; Singh, H. B.; Butcher, R. J. Chem. – Eur. J. 2011, 17, 12741–12755. doi:10.1002/chem.201100930 |
| 37. | Bhat, M. Y.; Kumar, A.; Ahmed, Q. N. Tetrahedron 2020, 76, 131105. doi:10.1016/j.tet.2020.131105 |
| 61. | Fernández Riveras, J. A.; Frontera, A.; Bauzá, A. Phys. Chem. Chem. Phys. 2021, 23, 17656–17662. doi:10.1039/d1cp01929e |
| 38. | Kim, H.; Ku, B.-C.; Goh, M.; Ko, H. C.; Ando, S.; You, N.-H. Macromolecules 2019, 52, 827–834. doi:10.1021/acs.macromol.8b02139 |
| 47. | Li, Q.; Zhang, J.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Polymers (Basel, Switz.) 2018, 10, 417. doi:10.3390/polym10040417 |
| 48. | Won, M.-S.; Yeom, J.-S.; Yoon, J.-H.; Jeong, E.-D.; Shim, Y.-B. Bull. Korean Chem. Soc. 2003, 24, 948–952. doi:10.5012/bkcs.2003.24.7.948 |
| 49. | Wen, Y.-H.; Li, X.-M.; Wang, L.; Zhang, S.-S. Acta Crystallogr., Sect. E: Struct. Rep. Online 2006, 62, o2185–o2186. doi:10.1107/s1600536806015972 |
| 46. | Tanini, D.; Dalia, C.; Capperucci, A. Green Chem. 2021, 23, 5680–5686. doi:10.1039/d0gc04322b |
| 37. | Bhat, M. Y.; Kumar, A.; Ahmed, Q. N. Tetrahedron 2020, 76, 131105. doi:10.1016/j.tet.2020.131105 |
| 43. | Dominic, J.; David, T.; Vanaja, A.; Satheesh Kumar, K. K. Eur. Polym. J. 2016, 85, 236–243. doi:10.1016/j.eurpolymj.2016.10.028 |
| 44. | Ibrahim, K. A. Arabian J. Chem. 2017, 10, S2668–S2674. doi:10.1016/j.arabjc.2013.10.010 |
| 45. | Manimegalai, P.; Dominic, J.; Satheesh Kumar, K. K. Polym. Int. 2022, 71, 770–776. |
| 37. | Bhat, M. Y.; Kumar, A.; Ahmed, Q. N. Tetrahedron 2020, 76, 131105. doi:10.1016/j.tet.2020.131105 |
| 41. | Ding, Y.; Liang, J.; Liu, G.; Ni, W.; Shen, L. Coatings 2019, 9, 399. doi:10.3390/coatings9060399 |
| 42. | Baker, A. G. ARO (Koya) 2019, 7, 47–52. doi:10.14500/aro.10483 |
© 2024 Selladurai and Karuthapandi; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.