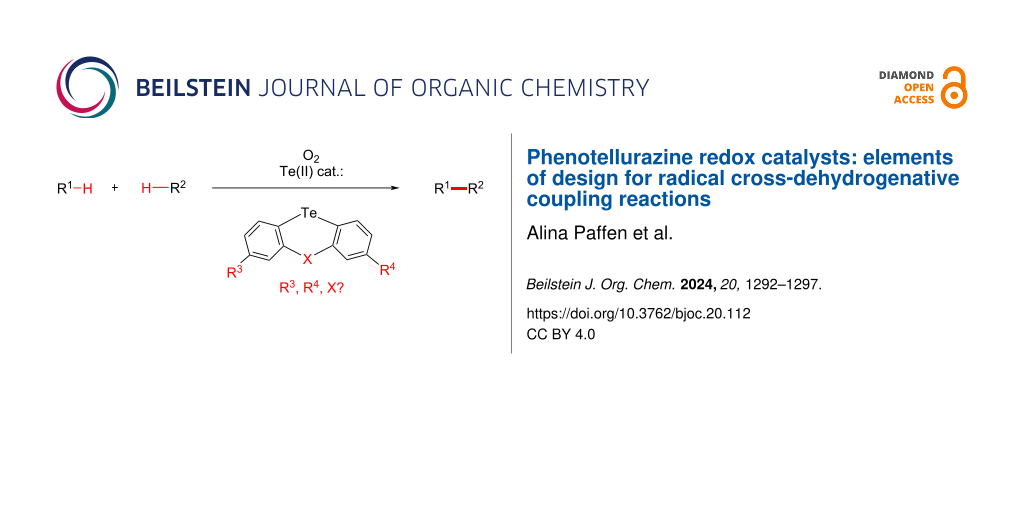
Abstract
Redox active phenotellurazine catalysts have been recently utilized in two different cross-dehydrogenative coupling reactions. In this study, we revisit the design of the phenotellurazine redox catalysts. In particular, we investigate the level of cooperativity between the Te- and N-centers, the effect of secondary versus tertiary N-centers, the effect of heterocyclic versus non-heterocyclic structures, and the effect of substitution patterns on the redox catalytic activity.

Graphical Abstract
Introduction
Tellurium catalysis has become increasingly important in recent years. This is due to its unique chalcogen bonding ability, thus enabling the activation of small yet highly relevant organic substrates. For example, Huber and co-authors recently designed a Te-based catalyst in an indole Michael addition reaction [1-5]. Pale and Mamane utilized another Te-based catalyst in an electrophilic bromine-mediated cyclization reaction [6,7], and Gabbaï yet another in a different cyclization reaction [8,9], among other catalytic chalcogen bonding activation examples [10-29]. In contrast, we have reported recently some redox-active Te-based catalysts, which exploit the redox flexibility of tellurium, especially in the context of phenotellurazine scaffolds. Notably, we showed that phenotellurazine PTeZ1 could significantly catalyze the cross-dehydrogenative phenothiazination of phenols bearing challenging electron-withdrawing substituents under a simple oxygen atmosphere (Scheme 1a) [30-32]. Most recently, we also showed that phenotellurazines could catalyze the oxidative dimerization of indoles, likewise under a simple oxygen atmosphere. 2-Methoxyphenotellurazine PTeZ2 proved to be the optimal catalyst in the latter case (Scheme 1b) [33].
![[1860-5397-20-112-i1]](/bjoc/content/inline/1860-5397-20-112-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Phenotellurazine-catalyzed cross-dehydrogenative couplings.
Scheme 1: Phenotellurazine-catalyzed cross-dehydrogenative couplings.
In the present study, we decided to revisit the design of the phenotellurazine redox catalyst, in the hope of improving it as well as enabling new catalytic reactivity. In particular, we wished to investigate and optimize the level of electronic cooperativity between the Te- and N-centers, the effect of secondary versus tertiary N-centers, the effect of heterocyclic versus non-heterocyclic structures, and the effect of various substitution patterns.
Results and Discussion
With this aim in mind, we thus started investigating Te(II) redox catalyst candidates that do not necessarily carry a N-atom in the structure, or else at different positions, in order to establish how their redox catalytic reactivity might be affected. Indeed, we learned recently that amino-arenes possess some level of redox catalytic activity by themselves, in the absence of a Te-center [34], and therefore wished to investigate and optimize their redox contribution within the Te(II) catalysts.
Multiple synthetic efforts were therefore deployed in order to access several key Te(II) targets, both with and without N-functional groups, in heterocyclic as well as in non-heterocyclic fashion. These were then investigated in the Te(II)-catalyzed benchmark dehydrogenative phenothiazination of phenols, a reaction that we discovered in 2015 [35-37], under analogous conditions as previously described [30-32]. The results are summarized in Scheme 2. The multistep synthesis and characterization of all Te-based catalyst candidates can be found in Supporting Information File 1. In the main article we will focus on catalysis results.
![[1860-5397-20-112-i2]](/bjoc/content/inline/1860-5397-20-112-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Screening of new Te(II)-catalyst candidates. ODCB: ortho-dichlorobenzene.
Scheme 2: Screening of new Te(II)-catalyst candidates. ODCB: ortho-dichlorobenzene.
We therefore first tested various telluracycle candidates, in particular without an NH bridge (catalyst candidates PTeZ3, Te4–Te7, Scheme 2A). These telluracycles, bridged by very diverse electron-donating or -withdrawing functional groups all featured significantly inferior catalytic activity in the selected benchmark reaction (O2-mediated dehydrogenative phenothiazination of 4-phenylphenol). It can therefore be deduced that the N-bridging atom is important for catalytic activity. In order to further investigate the matter, we next turned our attention to non-heterocyclic tellurethers Te8–Te13, bearing various substituents (Scheme 2B). Expectedly, simple diphenyl tellurether Te8 featured degraded catalytic activity (3aa, 55% after 3 h), although this still represents a significantly superior result compared to catalyst-free conditions (3aa, 22% after 3 h). Electron π-donating substituents considerably improved catalytic activity, with an optimum among tested structures for Te10 (3aa, 74% after 3 h), which is intriguingly similar to the previously reported best catalyst for this reaction: PTeZ1 (3aa, 73% after 3 h). In other words, the cyclic phenotellurazine character of the catalyst does not seem to be essential in the context of this particular reaction, as opposed to the ortho/para substitution pattern of Te- and N-atoms. Nevertheless, all results considered, we elected at this point to keep the cyclic phenotellurazine structure of the catalyst in the hope of increased catalytic robustness, especially in view of further increasing substitution. Next, we therefore tested methoxy-substituted PTeZ2, a successful catalyst structure which we recently developed for the cross-dehydrogenative coupling of indoles [33], in the same benchmark reaction. To our satisfaction, PTeZ2 proved to be the most active catalyst so far in this study (3aa, 99% after 3 h). In order to further optimize the catalyst structure, we then reduced the catalytic loading by one order of magnitude, from the standard 10 mol % to only 1 mol %, all other reaction parameters remaining identical. In these considerably more demanding conditions, PTeZ2 still performed admirably well (3aa, 45% at 3 h), significantly better than the non-catalyzed reaction (3aa, 22% after 3 h). Going from 2-methoxy towards 2,8-dimethoxy substitution (PTeZ15) allowed to significantly increase catalytic activity (3aa, 68% after 3 h), still at 1 mol % loading. Encouraged by this trend, which seemed to indicate that the more π-electron-donating substituents increase catalytic activity, we continued structural optimization. Thus, 2-dimethylamino-substituted PTeZ16 performed even better (3aa, 74% after 3 h and 1 mol % loading).
In order to evaluate these new Te-catalysts further, we then turned our attention to the second, arguably more challenging test reaction (Scheme 3). In particular, we hoped that the more π-electron-rich optimized PTeZ16 (2-dimethylamino substituent) would outperform previously published PTeZ2 (2-methoxy) in that particular reaction. Unfortunately, this was not the case. At 1 mol % catalytic loading and 8 h reaction time, PTeZ2 considerably outperforms PTeZ16 (indole 4a towards product 5a, 81 versus 41%, respectively). Intrigued by these results, we also checked yields of product 5a at only 3 h reaction time for both catalysts. Thus, at shorter reaction time, it transpires that PTeZ2 and PTeZ16 perform similarly overall (5a, 37 versus 40%, respectively). It can therefore be concluded that while PTeZ16 (2-dimethylamino) shows promising initial catalytic activity, it is not robust enough to survive the oxidative high temperature conditions for a prolonged period of time. In contrast, as was previously demonstrated in the literature, PTeZ2 (2-methoxy substituent) features a far greater chemical stability, to such an extent that it could be in large part recovered at the end of the reaction (see previous study) [33]. In other words, PTeZ2 (2-methoxy substituent) features the best compromise in terms of electronic effects, which affect both the stability and reactivity of the key catalytically active intermediate(s), possibly including chalcogen bonding activation ability of the substrates [38].
![[1860-5397-20-112-i3]](/bjoc/content/inline/1860-5397-20-112-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Phenotellurazine-catalyzed cross-dehydrogenative indole dimerization.
Scheme 3: Phenotellurazine-catalyzed cross-dehydrogenative indole dimerization.
Conclusion
In conclusion, we demonstrated the importance of the phenotellurazine scaffold, bearing both a Te(II) center as well as a N-bridge, for redox catalytic activity. However, although the idea of increasing the π-electron-rich character of the phenotellurazine catalyst had seemed very promising at first, our results show that this strategy leads to overall less robust Te(II) catalysts under oxidative reaction conditions. This in turn generally leads to inferior catalytic performance. Our future research efforts in the area of Te(II) catalysis will likely focus on milder coupling reactions on the one hand, and/or on novel more robust and more active ligand designs on the other. In particular, more investigations will likely be needed regarding the optimization of the possible Te-substrate interaction.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of synthesized compounds. | ||
| Format: PDF | Size: 3.2 MB | Download |
Acknowledgements
Pola A. Hild and Steffen Schauerte, both students at the RWTH Aachen University, are acknowledged for short internships in the group on topics peripherally related to this research.
Funding
DFG-funded transregional collaborative research center SFB/TRR 88 ‘‘Cooperative effects in homo and heterometallic complexes’’ (http://3MET.de) is acknowledged for generous financial support.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S. M. Angew. Chem., Int. Ed. 2019, 58, 16923–16927. doi:10.1002/anie.201910639
Return to citation in text: [1] -
Wonner, P.; Steinke, T.; Vogel, L.; Huber, S. M. Chem. – Eur. J. 2020, 26, 1258–1262. doi:10.1002/chem.201905057
Return to citation in text: [1] -
Tarannam, N.; Voelkel, M. H. H.; Huber, S. M.; Kozuch, S. J. Org. Chem. 2022, 87, 1661–1668. doi:10.1021/acs.joc.1c00894
Return to citation in text: [1] -
Steinke, T.; Wonner, P.; Gauld, R. M.; Heinrich, S.; Huber, S. M. Chem. – Eur. J. 2022, 28, e202200917. doi:10.1002/chem.202200917
Return to citation in text: [1] -
Pal, D.; Steinke, T.; Vogel, L.; Engelage, E.; Heinrich, S.; Kutzinski, D.; Huber, S. M. Adv. Synth. Catal. 2023, 365, 2718–2723. doi:10.1002/adsc.202300502
Return to citation in text: [1] -
Weiss, R.; Aubert, E.; Pale, P.; Mamane, V. Angew. Chem., Int. Ed. 2021, 60, 19281–19286. doi:10.1002/anie.202105482
Return to citation in text: [1] -
Groslambert, L.; Padilla-Hernandez, A.; Weiss, R.; Pale, P.; Mamane, V. Chem. – Eur. J. 2023, 29, e202203372. doi:10.1002/chem.202203372
Return to citation in text: [1] -
Zhou, B.; Gabbaï, F. P. J. Am. Chem. Soc. 2021, 143, 8625–8630. doi:10.1021/jacs.1c04482
Return to citation in text: [1] -
Zhou, B.; Gabbaï, F. P. Organometallics 2021, 40, 2371–2374. doi:10.1021/acs.organomet.1c00279
Return to citation in text: [1] -
Rettig, I. D.; Van, J.; Brauer, J. B.; Luo, W.; McCormick, T. M. Dalton Trans. 2019, 48, 5665–5673. doi:10.1039/c9dt00487d
Return to citation in text: [1] -
Lutkus, L. V.; Rettig, I. D.; Davies, K. S.; Hill, J. E.; Lohman, J. E.; Eskew, M. W.; Detty, M. R.; McCormick, T. M. Organometallics 2017, 36, 2588–2596. doi:10.1021/acs.organomet.7b00166
Return to citation in text: [1] -
Oba, M.; Tanaka, K.; Nishiyama, K.; Ando, W. J. Org. Chem. 2011, 76, 4173–4177. doi:10.1021/jo200496r
Return to citation in text: [1] -
Okada, Y.; Oba, M.; Arai, A.; Tanaka, K.; Nishiyama, K.; Ando, W. Inorg. Chem. 2010, 49, 383–385. doi:10.1021/ic9022745
Return to citation in text: [1] -
Oba, M.; Okada, Y.; Nishiyama, K.; Ando, W. Org. Lett. 2009, 11, 1879–1881. doi:10.1021/ol900240s
Return to citation in text: [1] -
You, Y.; Ahsan, K.; Detty, M. R. J. Am. Chem. Soc. 2003, 125, 4918–4927. doi:10.1021/ja029590m
Return to citation in text: [1] -
Ahsan, K.; Drake, M. D.; Higgs, D. E.; Wojciechowski, A. L.; Tse, B. N.; Bateman, M. A.; You, Y.; Detty, M. R. Organometallics 2003, 22, 2883–2890. doi:10.1021/om030232h
Return to citation in text: [1] -
Kanda, T.; Engman, L.; Cotgreave, I. A.; Powis, G. J. Org. Chem. 1999, 64, 8161–8169. doi:10.1021/jo990842k
Return to citation in text: [1] -
Detty, M. R.; Zhou, F.; Friedman, A. E. J. Am. Chem. Soc. 1996, 118, 313–318. doi:10.1021/ja953187g
Return to citation in text: [1] -
Engman, L.; Stern, D.; Pelcman, M.; Andersson, C. M. J. Org. Chem. 1994, 59, 1973–1979. doi:10.1021/jo00087a008
Return to citation in text: [1] -
Detty, M. R.; Friedman, A. E.; Oseroff, A. R. J. Org. Chem. 1994, 59, 8245–8250. doi:10.1021/jo00105a049
Return to citation in text: [1] -
Detty, M. R.; Gibson, S. L. Organometallics 1992, 11, 2147–2156. doi:10.1021/om00042a031
Return to citation in text: [1] -
Benz, S.; Poblador‐Bahamonde, A. I.; Low‐Ders, N.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 5408–5412. doi:10.1002/anie.201801452
Return to citation in text: [1] -
Nakamura, Y.; Yamago, S. Beilstein J. Org. Chem. 2013, 9, 1607–1612. doi:10.3762/bjoc.9.183
Return to citation in text: [1] -
Drake, M. D.; Bright, F. V.; Detty, M. R. J. Am. Chem. Soc. 2003, 125, 12558–12566. doi:10.1021/ja0367593
Return to citation in text: [1] -
McKee, D. W. Carbon 1984, 22, 513–516. doi:10.1016/0008-6223(84)90084-8
Return to citation in text: [1] -
Kolb, S.; Oliver, G. A.; Werz, D. B. Angew. Chem., Int. Ed. 2020, 59, 22306–22310. doi:10.1002/anie.202007314
Return to citation in text: [1] -
Haberhauer, G.; Gleiter, R. Angew. Chem., Int. Ed. 2020, 59, 21236–21243. doi:10.1002/anie.202010309
Return to citation in text: [1] -
Mehrparvar, S.; Wölper, C.; Gleiter, R.; Haberhauer, G. Org. Mater. 2022, 4, 43–52. doi:10.1055/a-1883-6076
Return to citation in text: [1] -
Mehrparvar, S.; Wölper, C.; Haberhauer, G. Angew. Chem., Int. Ed. 2023, 62, e202304202. doi:10.1002/anie.202304202
Return to citation in text: [1] -
Cremer, C.; Goswami, M.; Rank, C. K.; de Bruin, B.; Patureau, F. W. Angew. Chem., Int. Ed. 2021, 60, 6451–6456. doi:10.1002/anie.202015248
Return to citation in text: [1] [2] -
Cremer, C.; Eltester, M. A.; Bourakhouadar, H.; Atodiresei, I. L.; Patureau, F. W. Org. Lett. 2021, 23, 3243–3247. doi:10.1021/acs.orglett.1c00573
Return to citation in text: [1] [2] -
Vemuri, P. Y.; Cremer, C.; Patureau, F. W. Org. Lett. 2022, 24, 1626–1630. doi:10.1021/acs.orglett.2c00125
Return to citation in text: [1] [2] -
Cremer, C.; Patureau, F. W. JACS Au 2022, 2, 1318–1323. doi:10.1021/jacsau.2c00193
Return to citation in text: [1] [2] [3] -
Nandi, S.; Paffen, A.; Patureau, F. W. Synlett 2024, 35, 967–972. doi:10.1055/a-2225-8736
Return to citation in text: [1] -
Louillat‐Habermeyer, M.-L.; Jin, R.; Patureau, F. W. Angew. Chem., Int. Ed. 2015, 54, 4102–4104. doi:10.1002/anie.201500089
See for the original discovery of the dehydrogenative phenothiazination reaction.
Return to citation in text: [1] -
Goswami, M.; Konkel, A.; Rahimi, M.; Louillat‐Habermeyer, M.-L.; Kelm, H.; Jin, R.; de Bruin, B.; Patureau, F. W. Chem. – Eur. J. 2018, 24, 11936–11943. doi:10.1002/chem.201800730
See for the mechanistic investigation of the dehydrogenative phenothiazination reaction.
Return to citation in text: [1] -
Patureau, F. W. ChemCatChem 2019, 11, 5227–5231. doi:10.1002/cctc.201900152
See for an early review on the dehydrogenative phenothiazination reaction.
Return to citation in text: [1] -
Gleiter, R.; Haberhauer, G.; Werz, D. B.; Rominger, F.; Bleiholder, C. Chem. Rev. 2018, 118, 2010–2041. doi:10.1021/acs.chemrev.7b00449
Return to citation in text: [1]
| 1. | Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S. M. Angew. Chem., Int. Ed. 2019, 58, 16923–16927. doi:10.1002/anie.201910639 |
| 2. | Wonner, P.; Steinke, T.; Vogel, L.; Huber, S. M. Chem. – Eur. J. 2020, 26, 1258–1262. doi:10.1002/chem.201905057 |
| 3. | Tarannam, N.; Voelkel, M. H. H.; Huber, S. M.; Kozuch, S. J. Org. Chem. 2022, 87, 1661–1668. doi:10.1021/acs.joc.1c00894 |
| 4. | Steinke, T.; Wonner, P.; Gauld, R. M.; Heinrich, S.; Huber, S. M. Chem. – Eur. J. 2022, 28, e202200917. doi:10.1002/chem.202200917 |
| 5. | Pal, D.; Steinke, T.; Vogel, L.; Engelage, E.; Heinrich, S.; Kutzinski, D.; Huber, S. M. Adv. Synth. Catal. 2023, 365, 2718–2723. doi:10.1002/adsc.202300502 |
| 30. | Cremer, C.; Goswami, M.; Rank, C. K.; de Bruin, B.; Patureau, F. W. Angew. Chem., Int. Ed. 2021, 60, 6451–6456. doi:10.1002/anie.202015248 |
| 31. | Cremer, C.; Eltester, M. A.; Bourakhouadar, H.; Atodiresei, I. L.; Patureau, F. W. Org. Lett. 2021, 23, 3243–3247. doi:10.1021/acs.orglett.1c00573 |
| 32. | Vemuri, P. Y.; Cremer, C.; Patureau, F. W. Org. Lett. 2022, 24, 1626–1630. doi:10.1021/acs.orglett.2c00125 |
| 10. | Rettig, I. D.; Van, J.; Brauer, J. B.; Luo, W.; McCormick, T. M. Dalton Trans. 2019, 48, 5665–5673. doi:10.1039/c9dt00487d |
| 11. | Lutkus, L. V.; Rettig, I. D.; Davies, K. S.; Hill, J. E.; Lohman, J. E.; Eskew, M. W.; Detty, M. R.; McCormick, T. M. Organometallics 2017, 36, 2588–2596. doi:10.1021/acs.organomet.7b00166 |
| 12. | Oba, M.; Tanaka, K.; Nishiyama, K.; Ando, W. J. Org. Chem. 2011, 76, 4173–4177. doi:10.1021/jo200496r |
| 13. | Okada, Y.; Oba, M.; Arai, A.; Tanaka, K.; Nishiyama, K.; Ando, W. Inorg. Chem. 2010, 49, 383–385. doi:10.1021/ic9022745 |
| 14. | Oba, M.; Okada, Y.; Nishiyama, K.; Ando, W. Org. Lett. 2009, 11, 1879–1881. doi:10.1021/ol900240s |
| 15. | You, Y.; Ahsan, K.; Detty, M. R. J. Am. Chem. Soc. 2003, 125, 4918–4927. doi:10.1021/ja029590m |
| 16. | Ahsan, K.; Drake, M. D.; Higgs, D. E.; Wojciechowski, A. L.; Tse, B. N.; Bateman, M. A.; You, Y.; Detty, M. R. Organometallics 2003, 22, 2883–2890. doi:10.1021/om030232h |
| 17. | Kanda, T.; Engman, L.; Cotgreave, I. A.; Powis, G. J. Org. Chem. 1999, 64, 8161–8169. doi:10.1021/jo990842k |
| 18. | Detty, M. R.; Zhou, F.; Friedman, A. E. J. Am. Chem. Soc. 1996, 118, 313–318. doi:10.1021/ja953187g |
| 19. | Engman, L.; Stern, D.; Pelcman, M.; Andersson, C. M. J. Org. Chem. 1994, 59, 1973–1979. doi:10.1021/jo00087a008 |
| 20. | Detty, M. R.; Friedman, A. E.; Oseroff, A. R. J. Org. Chem. 1994, 59, 8245–8250. doi:10.1021/jo00105a049 |
| 21. | Detty, M. R.; Gibson, S. L. Organometallics 1992, 11, 2147–2156. doi:10.1021/om00042a031 |
| 22. | Benz, S.; Poblador‐Bahamonde, A. I.; Low‐Ders, N.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 5408–5412. doi:10.1002/anie.201801452 |
| 23. | Nakamura, Y.; Yamago, S. Beilstein J. Org. Chem. 2013, 9, 1607–1612. doi:10.3762/bjoc.9.183 |
| 24. | Drake, M. D.; Bright, F. V.; Detty, M. R. J. Am. Chem. Soc. 2003, 125, 12558–12566. doi:10.1021/ja0367593 |
| 25. | McKee, D. W. Carbon 1984, 22, 513–516. doi:10.1016/0008-6223(84)90084-8 |
| 26. | Kolb, S.; Oliver, G. A.; Werz, D. B. Angew. Chem., Int. Ed. 2020, 59, 22306–22310. doi:10.1002/anie.202007314 |
| 27. | Haberhauer, G.; Gleiter, R. Angew. Chem., Int. Ed. 2020, 59, 21236–21243. doi:10.1002/anie.202010309 |
| 28. | Mehrparvar, S.; Wölper, C.; Gleiter, R.; Haberhauer, G. Org. Mater. 2022, 4, 43–52. doi:10.1055/a-1883-6076 |
| 29. | Mehrparvar, S.; Wölper, C.; Haberhauer, G. Angew. Chem., Int. Ed. 2023, 62, e202304202. doi:10.1002/anie.202304202 |
| 8. | Zhou, B.; Gabbaï, F. P. J. Am. Chem. Soc. 2021, 143, 8625–8630. doi:10.1021/jacs.1c04482 |
| 9. | Zhou, B.; Gabbaï, F. P. Organometallics 2021, 40, 2371–2374. doi:10.1021/acs.organomet.1c00279 |
| 6. | Weiss, R.; Aubert, E.; Pale, P.; Mamane, V. Angew. Chem., Int. Ed. 2021, 60, 19281–19286. doi:10.1002/anie.202105482 |
| 7. | Groslambert, L.; Padilla-Hernandez, A.; Weiss, R.; Pale, P.; Mamane, V. Chem. – Eur. J. 2023, 29, e202203372. doi:10.1002/chem.202203372 |
| 30. | Cremer, C.; Goswami, M.; Rank, C. K.; de Bruin, B.; Patureau, F. W. Angew. Chem., Int. Ed. 2021, 60, 6451–6456. doi:10.1002/anie.202015248 |
| 31. | Cremer, C.; Eltester, M. A.; Bourakhouadar, H.; Atodiresei, I. L.; Patureau, F. W. Org. Lett. 2021, 23, 3243–3247. doi:10.1021/acs.orglett.1c00573 |
| 32. | Vemuri, P. Y.; Cremer, C.; Patureau, F. W. Org. Lett. 2022, 24, 1626–1630. doi:10.1021/acs.orglett.2c00125 |
| 33. | Cremer, C.; Patureau, F. W. JACS Au 2022, 2, 1318–1323. doi:10.1021/jacsau.2c00193 |
| 35. |
Louillat‐Habermeyer, M.-L.; Jin, R.; Patureau, F. W. Angew. Chem., Int. Ed. 2015, 54, 4102–4104. doi:10.1002/anie.201500089
See for the original discovery of the dehydrogenative phenothiazination reaction. |
| 36. |
Goswami, M.; Konkel, A.; Rahimi, M.; Louillat‐Habermeyer, M.-L.; Kelm, H.; Jin, R.; de Bruin, B.; Patureau, F. W. Chem. – Eur. J. 2018, 24, 11936–11943. doi:10.1002/chem.201800730
See for the mechanistic investigation of the dehydrogenative phenothiazination reaction. |
| 37. |
Patureau, F. W. ChemCatChem 2019, 11, 5227–5231. doi:10.1002/cctc.201900152
See for an early review on the dehydrogenative phenothiazination reaction. |
| 38. | Gleiter, R.; Haberhauer, G.; Werz, D. B.; Rominger, F.; Bleiholder, C. Chem. Rev. 2018, 118, 2010–2041. doi:10.1021/acs.chemrev.7b00449 |
| 34. | Nandi, S.; Paffen, A.; Patureau, F. W. Synlett 2024, 35, 967–972. doi:10.1055/a-2225-8736 |
| 33. | Cremer, C.; Patureau, F. W. JACS Au 2022, 2, 1318–1323. doi:10.1021/jacsau.2c00193 |
| 33. | Cremer, C.; Patureau, F. W. JACS Au 2022, 2, 1318–1323. doi:10.1021/jacsau.2c00193 |
© 2024 Paffen et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.