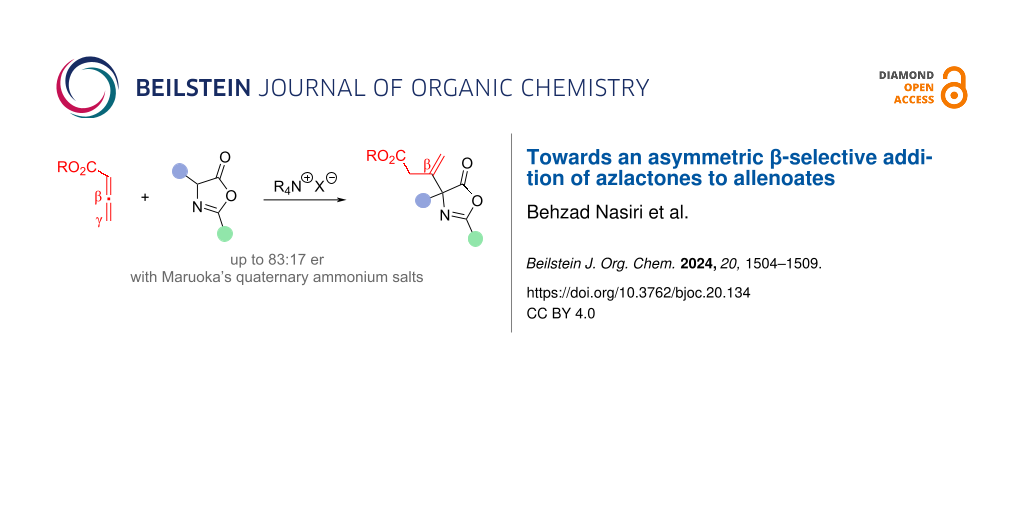
Abstract
We herein report the asymmetric organocatalytic addition of azlactones to allenoates. Upon using chiral quaternary ammonium salt catalysts, i.e., Maruoka’s binaphthyl-based spirocyclic ammonium salts, the addition of various azlactones to allenoates proceeds in a β-selective manner with moderate levels of enantioselectivities (up to 83:17 er). Furthermore, the obtained products can be successfully engaged in nucleophilic ring opening reactions, thus giving highly functionalized α-amino acid derivatives.

Graphical Abstract
Introduction
The development of asymmetric synthesis routes to access non-natural amino acids has for decades been one of the most heavily investigated tasks in organic synthesis and catalysis-oriented research [1-13]. As a consequence, a broad variety of conceptually orthogonal strategies to access differently functionalized non-natural α-amino acids (α-AA) [2-7] as well as β-amino acids (β-AA) [8-13] have been introduced and there is still considerable interest in the development of new concepts and synthesis approaches. Our group has a longstanding focus on the development of asymmetric organocatalytic methods to access non-natural chiral α- and β-AA [14-19]. Hereby we are especially interested in utilizing simple (prochiral) starting materials and carry out stereoselective α-functionalizations by reacting them with suited C- or heteroatom electrophiles. α-Amino acid-derived azlactones 1 are amongst the most commonly utilized starting materials to access more diverse chiral α,α-disubstituted amino acids (Scheme 1A) [20-22]. More specifically, these compounds can be engaged in a variety of asymmetric α-carbo- and α-heterofunctionalization reactions by utilizing different catalysis strategies [20-22]. We have recently carried out systematic investigations concerning the syntheses of advanced β-AA by means of asymmetric α-carbofunctionalization reactions and during these studies we also realized that the masked β-AA derivatives 2 undergo enantioselective β-addition to allenoates 3 under chiral ammonium salt catalysis (Scheme 1B) [18]. Interestingly, hereby we also found that the use of alternative catalyst systems (i.e., tertiary phosphines) allows for a γ-selective addition of 2 to the allenoate instead, thus resulting in two complementary catalyst-controlled pathways [18]. Based on these previous results, and also the well-documented different reactivity trends of allenoates 3 when using different organocatalysts and activation modes [23-27], we were thus wondering if we could extend this ammonium salt-catalyzed β-selective allenoate functionalization strategy to other amino acid classes. Azlactones 1 have previously been used for γ-selective additions to allenoates under chiral phosphine catalysis [28]. In addition, glycine Schiff base derivatives [29] as well as α-amino acid-based thiazolones [30] have successfully been used for asymmetric β-selective additions to allenoates when using chiral ammonium salt catalysts or chiral organobase catalysts. However, to the best of our knowledge the β-selective asymmetric addition of azlactones 1 to allenoates 3 delivering highly functionalized α,α-disubstituted α-amino acid derivatives 5 has so far not been systematically addressed (for recent other β-selective additions of enolate precursors to allenoates please see references [31-34]). Thus, we now became interested in testing this transformation under asymmetric ammonium salt catalysis [35-38] and the results of these investigations are outlined in this contribution (Scheme 1C).
![[1860-5397-20-134-i1]](/bjoc/content/inline/1860-5397-20-134-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General use of azlactones 1 to access more advanced α-AA derivatives (A), our recently reported ammonium salt-catalyzed β-selective addition of compounds 2 to allenoates 3 (B), and the herein investigated β-selective addition of azlactones 1 to allenoates 3 (C).
Scheme 1: General use of azlactones 1 to access more advanced α-AA derivatives (A), our recently reported amm...
Results and Discussion
We started our investigations by testing the quaternary ammonium salt-catalyzed addition of azlactone 1a to allenoate 3a (Table 1 gives an overview of the most significant results obtained hereby). First experiments using cinchona alkaloid-based quaternary ammonium salts A showed that the expected β-addition product 5a can be accessed under typical phase-transfer conditions, but with low selectivities and yields only when using these catalysts (Table 1, entries 1–4, other cinchona alkaloid-based ammonium salt derivatives as well as free base cinchona alkaloids were tested too but did not allow for any improvement). Using the established and commercially available Maruoka catalysts B1 and B2 [39] next turned out to be more promising (Table 1, entries 5–8). Testing the bis-CF3-substitued B1 first allowed for 75:25 er, but with moderate yield only when carrying out the reaction in toluene in the presence of 3 equiv of K2CO3 (Table 1, entry 5). Lower amounts of base (Table 1, entry 6) or other solvents, as exemplified for CH2Cl2 (Table 1, entry 7, similar non-selective results were obtained when using THF), were found to be less-suited however. Testing the 3,4,5-trifluorobenzene-decorated catalyst B2 with K2CO3 in toluene next (Table 1, entry 8) allowed for a slightly higher selectivity but still gave only a relatively low yield. Spirobiindane-based salts C emerged as promising alternative for quaternary ammonium salt scaffolds recently [40,41] and were also the catalysts of choice in our recently developed β-selective allenoate addition of isoxazolidinones 2 (compare with Scheme 1B [18]). Unfortunately, these catalysts were found to be less-suited for our azlactone protocol, as exemplified for derivative C1 (Table 1, entry 9). Accordingly, we carried out our final optimization using Maruoka’s catalyst B2 (Table 1, entries 10–14). By testing different bases and lower temperatures as well as lower catalyst loadings we identified the use of 3 equiv Cs2CO3 in toluene (0.05 M) at room temperature as the best-suited conditions (Table 1, entry 13), allowing for the synthesis of 5a in moderate yield (61%) and enantioselectivity (81:19 er).
Table 1: Optimization of the addition of azlactone 1a to allenoate 3aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-134-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Cat. | Base | Solvent | T [°C] | Yieldb | erc |
| 1 | A1 | K2CO3 | toluene | 25 | 41 | 58:42 |
| 2 | A2 | K2CO3 | toluene | 25 | 45 | 60:40 |
| 3 | A3 | K2CO3 | toluene | 25 | 40 | 58:42 |
| 4 | A4 | K2CO3 | toluene | 25 | 45 | 60:40 |
| 5 | B1 | K2CO3 | toluene | 25 | 55 | 75:25 |
| 6 | B1 | K2CO3 (1 equiv) | toluene | 25 | 20 | 72:28 |
| 7 | B1 | K2CO3 | CH2Cl2 | 25 | 33 | 51:49 |
| 8 | B2 | K2CO3 | toluene | 25 | 50 | 80:20 |
| 9 | C1 | K2CO3 | toluene | 25 | 40 | 68:32 |
| 10 | B2 | K2CO3 | toluene | 0 | 45 | 80:20 |
| 11 | B2 (5%) | K2CO3 | toluene | 0 | 41 | 77:23 |
| 12 | B2 | K3PO4 | toluene | 25 | 55 | 81:19 |
| 13 | B2 | Cs2CO3 | toluene | 25 | 61 | 81:19 |
| 14 | B2 | Cs2CO3 | toluene (0.1 M) | 25 | 75 | 73:27 |
aUnless otherwise stated, all reactions were carried out by stirring 1a (0.1 mmol), the allenoate (2 equiv), the indicated base and the catalyst, in the given solvent (0.05 M based on 1a) at the given temperature for 24 h. bIsolated yield. cDetermined by HPLC using a chiral stationary phase, (−)-5a was obtained as the major enantiomer when using the (R,R)-configurated catalysts B.
With optimized conditions for the synthesis of enantioenriched (−)-5a at hand, we next investigated the generality of this protocol. As outlined in Scheme 2, differently substituted allenoates were reasonably well tolerated (see products 5a–d), albeit some erosion in enantioselectivity was observed when using a tert-butyl ester containing allenoate (product 5d). Various α-arylmethyl-substituted azlactones 1 performed similarly as compared to the parent system 1a (products 5e–i), and analogous α-alkyl-substituted derivatives were reasonably well accepted too (5j–o). When varying the aryl substituent in position 2 of the oxazolone core (compare products 5a, 5g, and 5p) we found that increasing the steric bulk (5p) leads to a somewhat lower enantioselectivity, while the methoxy-substituent does not have a strong impact on the yield. It should, however, be stated that some of the methoxy-containing products, i.e., the α-alkyl-substituted 5j and 5k tend to undergo partial nucleophilic ring opening by residual water during column chromatography. Unfortunately, attempts to assign the absolute configuration of products 5 failed, as we have not been able to obtain any crystals suited for single crystal X-ray diffraction analysis.
![[1860-5397-20-134-i2]](/bjoc/content/inline/1860-5397-20-134-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Application scope (conditions as detailed in Table 1, entry 13).
Scheme 2: Application scope (conditions as detailed in Table 1, entry 13).
Finally, we also tested the suitability of products 5 to access acyclic α-AA derivatives by means of nucleophilic azlactone-opening reactions. Gratifyingly primary amines can be easily utilized under reflux conditions to access the amide derivatives 6a and 6b straightforwardly (Scheme 3), thus demonstrating the versatility of compounds 5 to access more complex acyclic α-AA derivatives in a straightforward manner.
![[1860-5397-20-134-i3]](/bjoc/content/inline/1860-5397-20-134-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
The development of novel catalytic methods for the asymmetric synthesis of non-natural amino acid derivatives is a contemporary task and we herein introduce an organocatalytic protocol for the β-selective addition of various azlactones 1 to allenoates 3. Upon using Maruoka’s spirocyclic binaphthyl-based quaternary ammonium salts B as catalysts this transformation can be achieved with enantioselectivities up to 83:17 er. Furthermore, the herein accessed cyclic products 5 could be successfully engaged in ring-opening reactions with different amines, thus giving access to the acyclic α-amino acid-based amides 6 straightforwardly.
Experimental
General details
1H and 13C NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer with a broad band observe probe. All NMR spectra were referenced on the solvent residual peak (CDCl3: δ 7.26 ppm for 1H NMR and δ 77.16 ppm for 13C NMR). NMR data are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet), coupling constants (Hz), relative integration value. High-resolution mass spectra were obtained using a Thermo Fisher Scientific LTQ Orbitrap XL spectrometer with an Ion Max API source and analyses were made in the positive ionization mode if not otherwise stated. Infrared (IR) spectra were recorded on a Bruker Alpha II FTIR spectrometer with diamond ATR-module using the OPUS software package and are reported in terms of frequency of absorption (cm−1). HPLC was performed using a Shimadzu Prominence system with a diode array detector with a CHIRALPAK AD-H, CHIRAL ART Amylose-SA, (250 × 4.6 mm, 5 µm) chiral stationary phase. Optical rotations were recorded on a Schmidt + Haensch Polarimeter Model UniPol L1000 at 589 nm ([α]D values are listed in deg/(dm(g/cm3)); concentration c is given in g/100 mL).
Unless otherwise stated, all chemicals were purchased from commercial suppliers and used without further purification. Dry solvents were obtained from an MBraun-SPS-800 solvent purification system. All reactions were carried out under argon atmosphere unless stated otherwise. Azlactones 1 and allenoates 3 were synthesized according to previously published procedures [18,42-44].
General procedure
An oven-dried Schlenk tube equipped with a stirring bar was charged with azlactone 1 (0.05–0.1 mmol), catalyst B2 (10 mol % related to 1), and Cs2CO3 (3 equiv). Then the respective allenoate 3 (2 equiv) and toluene (0.05 M with respect to 1) were added and the mixture was stirred at room temperature for 24 h (Ar atmosphere). The crude product was passed through a short column of silicagel (rinsed with DCM and EtOAc), concentrated under reduced pressure, and subsequently purified by preparative TLC (silica gel, heptanes/EtOAc 4:1) to obtain the products 2 in the given yields and enantiopurities.
Details for the parent compound 5a (details for the other targets can be found in Supporting Information File 1). Obtained as a colorless oil in 61% yield (81:19 er) on 0.1 mmol scale. [α]D22 = −11.4 (c 1.1, CHCl3); 1H NMR (300 MHz, CDCl3, 298.0 K) δ/ppm = 7.85 (dd, J = 8.6, 1.4 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.53 Hz, 2H), 7.24–7.11 (m, 5H), 5.79 (s, 1H), 5.37 (s, 1H), 4.14–3.90 (m, 2H), 3.52–3.16 (m, 4H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3, 298.0 K) δ/ppm = 177.4, 171.0, 160.3, 139.1, 133.8, 132.6, 130.5, 128.6, 128.0, 127.8, 127.3, 125.6, 118.1, 75.9, 60.9, 44.9, 39.3, 13.9; IR (neat): 3080, 3070, 2917, 1815, 1732, 1656, 1480, 1175, 1093, 1059, 1030, 974, 893, 694 cm−1; HRESIMS m/z: [C22H21NO4 + H]+ calcd for 364.1543; found, 364.1554; HPLC: (Chiralpak SA, eluent: n-hexane/iPrOH = 100:2, 0.5 mL·min−1, 20 °C, λ = 254 nm) retention times: tmajor = 16.15 min, tminor = 17.00 min.
Supporting Information
| Supporting Information File 1: Full experimental and analytical details and copies of NMR spectra and HPLC traces. | ||
| Format: PDF | Size: 3.6 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Hughes, A., Ed. Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1–5. doi:10.1002/9783527631766
Return to citation in text: [1] -
Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; American Chemical Society: Washington, DC, USA, 2009.
Return to citation in text: [1] [2] -
O’Donnell, M. J., Ed. α-Amino Acid Synthesis; Tetrahedron Symposia-in-Print, No. 33; Pergamon: Oxford, UK, 1988.
Return to citation in text: [1] [2] -
Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o
Return to citation in text: [1] [2] -
Metz, A. E.; Kozlowski, M. C. J. Org. Chem. 2015, 80, 1–7. doi:10.1021/jo502408z
Return to citation in text: [1] [2] -
Vogt, H.; Bräse, S. Org. Biomol. Chem. 2007, 5, 406–430. doi:10.1039/b611091f
Return to citation in text: [1] [2] -
Cativiela, C.; Ordonez, M. Tetrahedron: Asymmetry 2009, 20, 1–63. doi:10.1016/j.tetasy.2009.01.002
Return to citation in text: [1] [2] -
Juaristi, E.; López-Ruiz, H. Curr. Med. Chem. 1999, 6, 983–1004. doi:10.2174/092986730610220401161510
Return to citation in text: [1] [2] -
Abele, S.; Seebach, D. Eur. J. Org. Chem. 2000, 1–15. doi:10.1002/(sici)1099-0690(200001)2000:1<1::aid-ejoc1>3.0.co;2-6
Return to citation in text: [1] [2] -
Juaristi, E.; Soloshonok, V. A., Eds. Enantioselective Synthesis of b-Amino Acids, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. doi:10.1002/0471698482
Return to citation in text: [1] [2] -
Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656. doi:10.1039/b919599h
Return to citation in text: [1] [2] -
Ashfaq, M.; Tabassum, R.; Ahmad, M. M.; Hassan, N. A.; Oku, H.; Rivera, G. Med. Chem. 2015, 5, 295.
Return to citation in text: [1] [2] -
Noda, H.; Shibasaki, M. Eur. J. Org. Chem. 2020, 2350–2361. doi:10.1002/ejoc.201901596
Return to citation in text: [1] [2] -
Tiffner, M.; Novacek, J.; Busillo, A.; Gratzer, K.; Massa, A.; Waser, M. RSC Adv. 2015, 5, 78941–78949. doi:10.1039/c5ra14466c
Return to citation in text: [1] -
Eitzinger, A.; Winter, M.; Schörgenhumer, J.; Waser, M. Chem. Commun. 2020, 56, 579–582. doi:10.1039/c9cc09239k
Return to citation in text: [1] -
Zebrowski, P.; Eder, I.; Eitzinger, A.; Mallojjala, S. C.; Waser, M. ACS Org. Inorg. Au 2022, 2, 34–43. doi:10.1021/acsorginorgau.1c00025
Return to citation in text: [1] -
Haider, V.; Zebrowski, P.; Michalke, J.; Monkowius, U.; Waser, M. Org. Biomol. Chem. 2022, 20, 824–830. doi:10.1039/d1ob02235k
Return to citation in text: [1] -
Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493
Return to citation in text: [1] [2] [3] [4] [5] -
Stockhammer, L.; Craik, R.; Monkowius, U.; Cordes, D. B.; Smith, A. D.; Waser, M. ChemistryEurope 2023, 1, e202300015. doi:10.1002/ceur.202300015
Return to citation in text: [1] -
Alba, A.-N. R.; Rios, R. Chem. – Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636
Return to citation in text: [1] [2] -
de Castro, P. P.; Carpanez, A. G.; Amarante, G. W. Chem. – Eur. J. 2016, 22, 10294–10318. doi:10.1002/chem.201600071
Return to citation in text: [1] [2] -
Marra, I. F. S.; de Castro, P. P.; Amarante, G. W. Eur. J. Org. Chem. 2019, 5830–5855. doi:10.1002/ejoc.201901076
Return to citation in text: [1] [2] -
Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x
Return to citation in text: [1] -
Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102. doi:10.1039/b816700c
Return to citation in text: [1] -
Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460
Return to citation in text: [1] -
Fan, Y. C.; Kwon, O. Chem. Commun. 2013, 49, 11588. doi:10.1039/c3cc47368f
Return to citation in text: [1] -
Wang, Z.; Xu, X.; Kwon, O. Chem. Soc. Rev. 2014, 43, 2927–2940. doi:10.1039/c4cs00054d
Return to citation in text: [1] -
Wang, T.; Yu, Z.; Hoon, D. L.; Phee, C. Y.; Lan, Y.; Lu, Y. J. Am. Chem. Soc. 2016, 138, 265–271. doi:10.1021/jacs.5b10524
Return to citation in text: [1] -
Elsner, P.; Bernardi, L.; Salla, G. D.; Overgaard, J.; Jørgensen, K. A. J. Am. Chem. Soc. 2008, 130, 4897–4905. doi:10.1021/ja710689c
Return to citation in text: [1] -
Uraguchi, D.; Kawai, Y.; Sasaki, H.; Yamada, K.; Ooi, T. Chem. Lett. 2018, 47, 594–597. doi:10.1246/cl.180031
Return to citation in text: [1] -
Shu, L.; Wang, P.; Gu, C.; Liu, W.; Alabanza, L. M.; Zhang, Y. Org. Process Res. Dev. 2013, 17, 651–657. doi:10.1021/op300306c
Return to citation in text: [1] -
Jin, N.; Misaki, T.; Sugimura, T. Chem. Lett. 2013, 42, 894–896. doi:10.1246/cl.130295
Return to citation in text: [1] -
Vaishanv, N. K.; Zaheer, M. K.; Kant, R.; Mohanan, K. Eur. J. Org. Chem. 2019, 6138–6142. doi:10.1002/ejoc.201901199
Return to citation in text: [1] -
Liu, Y.-L.; Wang, X.-P.; Wei, J.; Li, Y. Tetrahedron 2022, 103, 132577. doi:10.1016/j.tet.2021.132577
Return to citation in text: [1] -
Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. doi:10.1002/anie.201206835
Return to citation in text: [1] -
Qian, D.; Sun, J. Chem. – Eur. J. 2019, 25, 3740–3751. doi:10.1002/chem.201803752
Return to citation in text: [1] -
Albanese, D. C. M.; Penso, M. Eur. J. Org. Chem. 2023, 26, 10.1002/ejoc.202300224. doi:10.1002/ejoc.202300224
Return to citation in text: [1] -
Otevrel, J.; Waser, M. Asymmetric Phase-Transfer Catalysis- From Classical Applications to New Concepts. In Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities; Albrecht, L.; Albrecht, A.; Dell'Amico, L., Eds.; Wiley-VCH: Weinheim, Germany, 2023; pp 71–120. doi:10.1002/9783527832217.ch3
Return to citation in text: [1] -
Lee, H.-J.; Maruoka, K. Chem. Rec. 2023, 23, e202200286. doi:10.1002/tcr.202200286
Return to citation in text: [1] -
Xu, C.; Qi, Y.; Yang, X.; Li, X.; Li, Z.; Bai, L. Org. Lett. 2021, 23, 2890–2894. doi:10.1021/acs.orglett.1c00535
Return to citation in text: [1] -
Xu, C.; Yang, X. Synlett 2022, 33, 664–668. doi:10.1055/a-1795-7740
Return to citation in text: [1] -
Macovei, C.; Vicennati, P.; Quinton, J.; Nevers, M.-C.; Volland, H.; Créminon, C.; Taran, F. Chem. Commun. 2012, 48, 4411. doi:10.1039/c2cc31312j
Return to citation in text: [1] -
de Mello, A. C.; Momo, P. B.; Burtoloso, A. C. B.; Amarante, G. W. J. Org. Chem. 2018, 83, 11399–11406. doi:10.1021/acs.joc.8b01683
Return to citation in text: [1] -
Žabka, M.; Kocian, A.; Bilka, S.; Andrejčák, S.; Šebesta, R. Eur. J. Org. Chem. 2019, 6077–6087. doi:10.1002/ejoc.201901052
Return to citation in text: [1]
| 18. | Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493 |
| 18. | Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493 |
| 42. | Macovei, C.; Vicennati, P.; Quinton, J.; Nevers, M.-C.; Volland, H.; Créminon, C.; Taran, F. Chem. Commun. 2012, 48, 4411. doi:10.1039/c2cc31312j |
| 43. | de Mello, A. C.; Momo, P. B.; Burtoloso, A. C. B.; Amarante, G. W. J. Org. Chem. 2018, 83, 11399–11406. doi:10.1021/acs.joc.8b01683 |
| 44. | Žabka, M.; Kocian, A.; Bilka, S.; Andrejčák, S.; Šebesta, R. Eur. J. Org. Chem. 2019, 6077–6087. doi:10.1002/ejoc.201901052 |
| 1. | Hughes, A., Ed. Amino Acids, Peptides and Proteins in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2009; Vol. 1–5. doi:10.1002/9783527631766 |
| 2. | Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; American Chemical Society: Washington, DC, USA, 2009. |
| 3. | O’Donnell, M. J., Ed. α-Amino Acid Synthesis; Tetrahedron Symposia-in-Print, No. 33; Pergamon: Oxford, UK, 1988. |
| 4. | Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 5. | Metz, A. E.; Kozlowski, M. C. J. Org. Chem. 2015, 80, 1–7. doi:10.1021/jo502408z |
| 6. | Vogt, H.; Bräse, S. Org. Biomol. Chem. 2007, 5, 406–430. doi:10.1039/b611091f |
| 7. | Cativiela, C.; Ordonez, M. Tetrahedron: Asymmetry 2009, 20, 1–63. doi:10.1016/j.tetasy.2009.01.002 |
| 8. | Juaristi, E.; López-Ruiz, H. Curr. Med. Chem. 1999, 6, 983–1004. doi:10.2174/092986730610220401161510 |
| 9. | Abele, S.; Seebach, D. Eur. J. Org. Chem. 2000, 1–15. doi:10.1002/(sici)1099-0690(200001)2000:1<1::aid-ejoc1>3.0.co;2-6 |
| 10. | Juaristi, E.; Soloshonok, V. A., Eds. Enantioselective Synthesis of b-Amino Acids, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. doi:10.1002/0471698482 |
| 11. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656. doi:10.1039/b919599h |
| 12. | Ashfaq, M.; Tabassum, R.; Ahmad, M. M.; Hassan, N. A.; Oku, H.; Rivera, G. Med. Chem. 2015, 5, 295. |
| 13. | Noda, H.; Shibasaki, M. Eur. J. Org. Chem. 2020, 2350–2361. doi:10.1002/ejoc.201901596 |
| 20. | Alba, A.-N. R.; Rios, R. Chem. – Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636 |
| 21. | de Castro, P. P.; Carpanez, A. G.; Amarante, G. W. Chem. – Eur. J. 2016, 22, 10294–10318. doi:10.1002/chem.201600071 |
| 22. | Marra, I. F. S.; de Castro, P. P.; Amarante, G. W. Eur. J. Org. Chem. 2019, 5830–5855. doi:10.1002/ejoc.201901076 |
| 39. | Lee, H.-J.; Maruoka, K. Chem. Rec. 2023, 23, e202200286. doi:10.1002/tcr.202200286 |
| 14. | Tiffner, M.; Novacek, J.; Busillo, A.; Gratzer, K.; Massa, A.; Waser, M. RSC Adv. 2015, 5, 78941–78949. doi:10.1039/c5ra14466c |
| 15. | Eitzinger, A.; Winter, M.; Schörgenhumer, J.; Waser, M. Chem. Commun. 2020, 56, 579–582. doi:10.1039/c9cc09239k |
| 16. | Zebrowski, P.; Eder, I.; Eitzinger, A.; Mallojjala, S. C.; Waser, M. ACS Org. Inorg. Au 2022, 2, 34–43. doi:10.1021/acsorginorgau.1c00025 |
| 17. | Haider, V.; Zebrowski, P.; Michalke, J.; Monkowius, U.; Waser, M. Org. Biomol. Chem. 2022, 20, 824–830. doi:10.1039/d1ob02235k |
| 18. | Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493 |
| 19. | Stockhammer, L.; Craik, R.; Monkowius, U.; Cordes, D. B.; Smith, A. D.; Waser, M. ChemistryEurope 2023, 1, e202300015. doi:10.1002/ceur.202300015 |
| 40. | Xu, C.; Qi, Y.; Yang, X.; Li, X.; Li, Z.; Bai, L. Org. Lett. 2021, 23, 2890–2894. doi:10.1021/acs.orglett.1c00535 |
| 41. | Xu, C.; Yang, X. Synlett 2022, 33, 664–668. doi:10.1055/a-1795-7740 |
| 8. | Juaristi, E.; López-Ruiz, H. Curr. Med. Chem. 1999, 6, 983–1004. doi:10.2174/092986730610220401161510 |
| 9. | Abele, S.; Seebach, D. Eur. J. Org. Chem. 2000, 1–15. doi:10.1002/(sici)1099-0690(200001)2000:1<1::aid-ejoc1>3.0.co;2-6 |
| 10. | Juaristi, E.; Soloshonok, V. A., Eds. Enantioselective Synthesis of b-Amino Acids, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. doi:10.1002/0471698482 |
| 11. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656. doi:10.1039/b919599h |
| 12. | Ashfaq, M.; Tabassum, R.; Ahmad, M. M.; Hassan, N. A.; Oku, H.; Rivera, G. Med. Chem. 2015, 5, 295. |
| 13. | Noda, H.; Shibasaki, M. Eur. J. Org. Chem. 2020, 2350–2361. doi:10.1002/ejoc.201901596 |
| 31. | Shu, L.; Wang, P.; Gu, C.; Liu, W.; Alabanza, L. M.; Zhang, Y. Org. Process Res. Dev. 2013, 17, 651–657. doi:10.1021/op300306c |
| 32. | Jin, N.; Misaki, T.; Sugimura, T. Chem. Lett. 2013, 42, 894–896. doi:10.1246/cl.130295 |
| 33. | Vaishanv, N. K.; Zaheer, M. K.; Kant, R.; Mohanan, K. Eur. J. Org. Chem. 2019, 6138–6142. doi:10.1002/ejoc.201901199 |
| 34. | Liu, Y.-L.; Wang, X.-P.; Wei, J.; Li, Y. Tetrahedron 2022, 103, 132577. doi:10.1016/j.tet.2021.132577 |
| 2. | Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; American Chemical Society: Washington, DC, USA, 2009. |
| 3. | O’Donnell, M. J., Ed. α-Amino Acid Synthesis; Tetrahedron Symposia-in-Print, No. 33; Pergamon: Oxford, UK, 1988. |
| 4. | Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 5. | Metz, A. E.; Kozlowski, M. C. J. Org. Chem. 2015, 80, 1–7. doi:10.1021/jo502408z |
| 6. | Vogt, H.; Bräse, S. Org. Biomol. Chem. 2007, 5, 406–430. doi:10.1039/b611091f |
| 7. | Cativiela, C.; Ordonez, M. Tetrahedron: Asymmetry 2009, 20, 1–63. doi:10.1016/j.tetasy.2009.01.002 |
| 35. | Shirakawa, S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 4312–4348. doi:10.1002/anie.201206835 |
| 36. | Qian, D.; Sun, J. Chem. – Eur. J. 2019, 25, 3740–3751. doi:10.1002/chem.201803752 |
| 37. | Albanese, D. C. M.; Penso, M. Eur. J. Org. Chem. 2023, 26, 10.1002/ejoc.202300224. doi:10.1002/ejoc.202300224 |
| 38. | Otevrel, J.; Waser, M. Asymmetric Phase-Transfer Catalysis- From Classical Applications to New Concepts. In Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities; Albrecht, L.; Albrecht, A.; Dell'Amico, L., Eds.; Wiley-VCH: Weinheim, Germany, 2023; pp 71–120. doi:10.1002/9783527832217.ch3 |
| 23. | Lu, X.; Zhang, C.; Xu, Z. Acc. Chem. Res. 2001, 34, 535–544. doi:10.1021/ar000253x |
| 24. | Cowen, B. J.; Miller, S. J. Chem. Soc. Rev. 2009, 38, 3102. doi:10.1039/b816700c |
| 25. | Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460 |
| 26. | Fan, Y. C.; Kwon, O. Chem. Commun. 2013, 49, 11588. doi:10.1039/c3cc47368f |
| 27. | Wang, Z.; Xu, X.; Kwon, O. Chem. Soc. Rev. 2014, 43, 2927–2940. doi:10.1039/c4cs00054d |
| 29. | Elsner, P.; Bernardi, L.; Salla, G. D.; Overgaard, J.; Jørgensen, K. A. J. Am. Chem. Soc. 2008, 130, 4897–4905. doi:10.1021/ja710689c |
| 18. | Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493 |
| 30. | Uraguchi, D.; Kawai, Y.; Sasaki, H.; Yamada, K.; Ooi, T. Chem. Lett. 2018, 47, 594–597. doi:10.1246/cl.180031 |
| 18. | Zebrowski, P.; Röser, K.; Chrenko, D.; Pospíšil, J.; Waser, M. Synthesis 2023, 55, 1706–1713. doi:10.1055/a-1948-5493 |
| 20. | Alba, A.-N. R.; Rios, R. Chem. – Asian J. 2011, 6, 720–734. doi:10.1002/asia.201000636 |
| 21. | de Castro, P. P.; Carpanez, A. G.; Amarante, G. W. Chem. – Eur. J. 2016, 22, 10294–10318. doi:10.1002/chem.201600071 |
| 22. | Marra, I. F. S.; de Castro, P. P.; Amarante, G. W. Eur. J. Org. Chem. 2019, 5830–5855. doi:10.1002/ejoc.201901076 |
| 28. | Wang, T.; Yu, Z.; Hoon, D. L.; Phee, C. Y.; Lan, Y.; Lu, Y. J. Am. Chem. Soc. 2016, 138, 265–271. doi:10.1021/jacs.5b10524 |
© 2024 Nasiri et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.