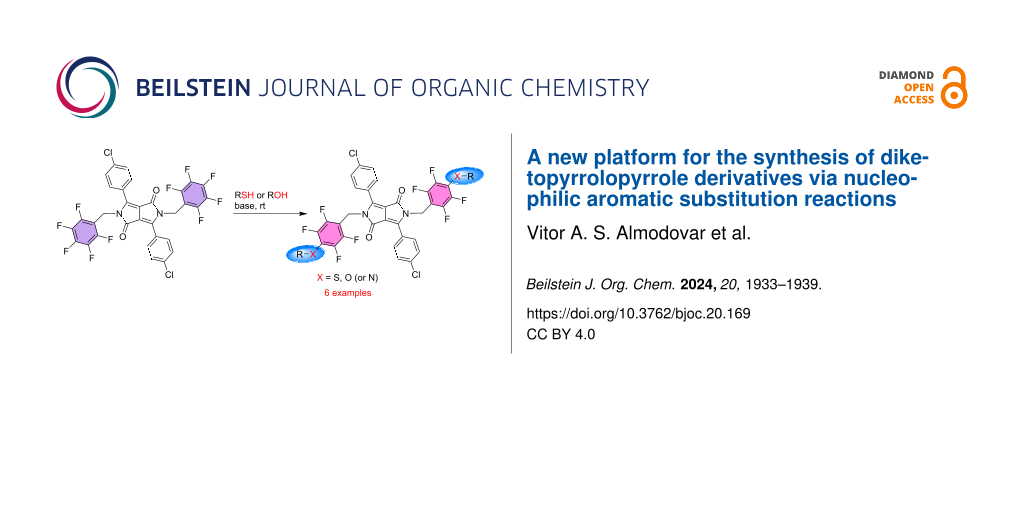
Abstract
Diketopyrrolopyrroles (DPPs) are a versatile group of dyes and pigments with valuable optoelectronic properties. In this work we report the synthesis of highly fluorescent DPP derivatives through straightforward nucleophilic aromatic substitution reactions with thiols and phenols. These nucleophilic substitutions occur at room temperature and manifest a remarkable selectivity for the 4-position of the pentafluorophenyl groups. Both symmetrical (disubstitution) and non-symmetrical (monosubstitution) DPP derivatives are formed in excellent overall yields. The optical properties of the newly synthesized compounds are also discussed. The new platform may be useful for bioorthogonal chemistry.

Graphical Abstract
Introduction
Diketopyrrolopyrroles (DPPs) are a class of organic pigments discovered by serendipity in the 1970s [1,2]. Generally, N-unsubstituted DPP derivatives exhibit high melting points, low solubility in most solvents, and strong absorption in the visible region [3,4]. In turn, N-substituted DPP derivatives are soluble in common organic solvents, exhibit large molar extinction coefficients, Stokes shifts in the range of 10–70 nm and high fluorescence quantum yields [5-7].
Due to their outstanding photophysical properties, DPP-based dyes have been used in a wide range of applications, namely as organic semiconductors [8], acceptors for organic solar cells [9,10], as fluorescent probes [11-13], or as photosensitizers for photodynamic therapy and antimicrobial photodynamic therapy [14-17]. DPP derivatives with improved performance or novel properties can be prepared by conventional chemical modifications of simple DPP derivatives [3,18]. The most frequently used transformations include: i) N-alkylation with adequately functionalized alkyl groups [19-22], ii) N-arylation [23-25], and functionalization at the 3,6-di(het)aryl groups via Suzuki–Miyaura [26-28] or Sonogashira [29-31] reactions.
In this study, we report a straightforward method to obtain a diverse array of N-substituted DPP derivatives through a two-step process. Firstly, the N-alkylation of Pigment Red 254 (DPP 1) is achieved using pentafluorobenzyl bromide, followed by a nucleophilic aromatic substitution (SNAr) with thiols and phenols. This approach is based on the well-established reactivity of perfluoroaromatic compounds in nucleophilic aromatic substitutions [32-35]. By varying the reaction conditions and the number of equivalents of the nucleophile, it is possible to promote the substitution of one or more fluorine atoms. Nucleophilic substitution of fluorine atoms often necessitates harsh conditions such as elevated temperatures, strong bases, or strong nucleophiles, but our findings demonstrate that this process can be conducted under remarkably mild conditions.
Results and Discussion
The initial step of our method involved the N-alkylation of DPP 1 with pentafluorobenzyl bromide (Scheme 1). Although a similar reaction had been previously reported for other DPP derivatives, the experimental conditions used (DMF, K2CO3, 120 °C, 2 h) resulted in very low yields (6–16%) for the formation of N,N’-bis(pentafluorobenzyl)-DPP derivatives [36]. Changing the base to NaH and performing the reaction at a lower temperature, enabled to obtain DPP 2 in a reasonable yield (61%) and allowed us to use it as a starting material for generating new DPP derivatives through nucleophilic aromatic substitution reactions with thiols and phenols.
![[1860-5397-20-169-i1]](/bjoc/content/inline/1860-5397-20-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of new diketopyrrolopyrroles via nucleophilic aromatic substitution.
Scheme 1: Synthesis of new diketopyrrolopyrroles via nucleophilic aromatic substitution.
The main objective of this study was to employ the N,N’-bis(pentafluorobenzyl)-DPP 2 as an electrophile and investigate its reactivity with thiols and phenols (Scheme 1). All SNAr reactions were performed in dry DMF at room temperature, in the presence of a base (K2CO3 or Cs2CO3). Room temperature was chosen due to the observed rapid degradation of the starting material at elevated temperatures. The work described herein allowed us to assess the potential of DPP 2 as a novel platform for obtaining functionalized DPP derivatives. As anticipated, it displayed reactivity with thiols and phenols through nucleophilic aromatic substitution at the pentafluorobenzyl groups, yielding both symmetrical (disubstitution) and non-symmetrical (monosubstitution) derivatives in satisfactory yields (Scheme 1).
Thiols are excellent nucleophiles and generally react under mild conditions, resulting in the substitution of the 4-F atom of the pentafluorophenyl groups. In this case, reactions with thiols were performed in dry DMF and K2CO3 was used as the base. Three different thiols were tested: pyridine-4-thiol, pyridine-2-thiol and 4-(acetylamino)benzenethiol. The reaction with pyridine-4-thiol yielded a mixture of the di- and monosubstituted compounds 3a and 4a in 51% and 23% yields, respectively. Conversely, for the reaction with pyridine-2-thiol, exclusively produced the disubstituted compound 3b in an 85% yield. Furthermore, the reaction with 4-(acetylamino)benzenethiol led to the selective formation of the disubstituted compound 3c in 53% yield.
Phenols are less nucleophilic than thiols and, depending on the substitution pattern, a stronger base is often required to generate the corresponding alkoxide, which is the effective nucleophile. So, in this case, Cs2CO3 was employed as the base. The reaction of DPP 2 with methyl 4-hydroxybenzoate yielded compounds 3d and 4d in 56% and 14% yield, respectively. When reacting with 4-(2,4,4-trimethylpentan-2-yl)phenol, the disubstituted compound 3e was obtained in 63% yield. In contrast to the reaction with pyridine-4-thiol, which resulted in the S-substituted product 3a, the reaction with 4-hydroxypyridine led exclusively to the formation of the pyridin-4-one-derived compounds 3f and 4f, in 45% and 13% yield, respectively. The substitution occurred at the nitrogen atom rather than the oxygen due to the preferential existence of 4-hydroxypyridine in the pyridin-4-one tautomeric form [37-39]. The structures of dyes 3a–f, 4a, 4d and 4f were unambiguously established through their 1H, 13C and 19F NMR and mass spectra.
The 1H NMR spectra of the symmetrical compounds displayed a characteristic signal for the N–CH2 protons as a singlet at approximately δ 5.10 ppm. Signals of the 4-chlorophenyl groups appeared as AB systems centred at around δ 7.9 ppm. For the non-symmetrical derivatives, two singlets were observed at approximately δ 5.05 and 5.10 ppm, corresponding to the protons of the N–CH2C6F5 and N–CH2C6F4XR groups, respectively. All 19F NMR spectra confirmed the selective substitution of the 4-fluorine atoms (in one or in two rings) by the disappearance of the signal corresponding to the resonance of those atoms. Mass spectra of compounds 3a–f, 4a, 4d and 4f consistently displayed the protonated molecular ion [M + H]+ as the base peak.
The UV–vis and fluorescence spectra of DPP derivatives 3a–f, 4a, 4d and 4f in DMF are presented in Figure 1, and their photophysical properties are summarized in Table 1. These compounds are highly fluorescent, and their UV–vis spectra are very similar. These results indicate that substituents with different functional groups can be attached to DPP 2 without significant modification of their optical properties. The observed Stokes shifts for dyes 3 and 4 averaged in the range of 60–70 nm. All compounds exhibited high fluorescence quantum yields, ranging from 0.66 to 0.83, confirming their potential applications in fluorescence imaging, sensors, and optoelectronic devices. A comprehensive discussion of the potential uses of these fluorescent substances in areas such as materials science, biology, or chemistry may provide a deeper understanding of their significance.
![[1860-5397-20-169-1]](/bjoc/content/figures/1860-5397-20-169-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (A) Absorption and (B) fluorescence spectra of compounds 3a–f, 4a, 4d and 4f, in DMF. Different concentrations of the compounds were used to allow visualization of each spectrum.
Figure 1: (A) Absorption and (B) fluorescence spectra of compounds 3a–f, 4a, 4d and 4f, in DMF. Different con...
Table 1: Spectroscopic data for the new compounds (between 1 × 10–6 M and 4 × 10–5 M in DMF).
| Compound | Absorption λmax (nm) | Log ε (M–1 cm–1) | Emission λmax (nm) | Stokes shift (cm–1) | ФF (DMF)a |
| 3a | 460 | 4.15 | 521 | 2545 | 0.69 |
| 4a | 460 | 4.11 | 522 | 2582 | 0.68 |
| 3b | 461 | 4.17 | 524 | 2608 | 0.83 |
| 3c | 461 | 4.25 | 525 | 2644 | 0.78 |
| 3d | 457 | 4.43 | 522 | 2687 | 0.73 |
| 4d | 459 | 4.46 | 521 | 2593 | 0.72 |
| 3e | 460 | 4.03 | 522 | 2547 | 0.71 |
| 3f | 456 | 4.33 | 523 | 2809 | 0.83 |
| 4f | 458 | 4.32 | 524 | 2750 | 0.66 |
aExcitation at 436 nm. N,N’-Dibenzyl-DPP was used as the fluorescence quantum yield reference: ΦF = 0.88, in chloroform [40].
Conclusion
In conclusion, novel DPP derivatives were synthesized through the reaction of a N,N’-bis(pentafluorobenzyl)-DPP with thiols and phenols. The nucleophilic aromatic substitution reactions took place under exceptionally mild experimental conditions, and the resulting compounds were isolated in reasonable yields. The newly synthesized compounds display high fluorescence quantum yields and moderate Stokes shifts, which are crucial attributes for their potential application in diverse fields, particularly in biological or technical applications. Additionally, it is crucial to highlight the chemical versatility of compound 2, which allows the attachment of various functional units without significantly altering its optical properties. This versatility holds significant promise in the design and synthesis of innovative molecules tailored for specific purposes. This study not only contributes to the expansion of accessible N-substituted DPP derivatives but also reveals that such transformations can be achieved with outstanding efficiency and environmental sensitivity by employing mild reaction conditions.
Experimental
Chemicals and instrumentation
The reagents used in this work were purchased from Merck Life Science (Algés, Portugal) or TCI Europe N.V. (Belgium) and were used as received. Pigment Red 254 was purchased from TCI Europe N.V. The solvents were used as received or distilled and dried by standard procedures. Analytical thin-layer chromatography (TLC) was carried out on precoated sheets with silica gel (Merck 60, 0.2 mm thick). Preparative TLC was carried out on 20 cm × 20 cm glass plates precoated with a layer of silica gel 60 (0.5 mm thick) and activated in an oven at 100 °C for 12 h. Melting points were determined with a Büchi B-540 apparatus. NMR spectra were recorded on a Bruker DRX 300 Avance operating at 300.13 MHz (for 1H NMR), at 75.47 MHz (for 13C NMR) and 282 MHz (for 19F NMR). Deuterated chloroform (CDCl3) was used as the solvent and tetramethylsilane (TMS) as the internal reference. The chemical shifts (δ) are expressed in parts per million (ppm) and the coupling constants (J) in hertz (Hz). UV–vis spectra were recorded on a Shimadzu UV-2501PC spectrophotometer using DMF as the solvent. The emission spectra were recorded with a Jasco FP-8300 spectrofluorometer using DMF as the solvent. Mass spectra were recorded using a Micromass Q-TOF-2TM mass spectrometer and CHCl3 as the solvent. The NMR, absorption and emission spectra of the new compounds are shown in Supporting Information File 1.
Synthesis
3,6-Bis(4-chlorophenyl)-2,5-bis(pentafluorobenzyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (2)
A suspension of DPP 1 (1 g, 2.8 mmol) and NaH (11.2 mmol) in DMF (60 mL) was stirred at 0 °C under a nitrogen atmosphere for 30 min. At this temperature, and under vigorous stirring, a solution of pentafluorobenzyl bromide (1.7 mL, 11.2 mmol) in DMF (8 mL) was added dropwise. The mixture was stirred for 24 h at room temperature and then it was diluted with CH2Cl2 and water. The organic layer was separated and washed with water and brine. The product was isolated by column chromatography on silica gel using CH2Cl2 as the eluent. Yield: 61%; mp: 278–280 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 7.62–7.66 (m, 4H), 7.47–7.52 (m, 4H), 5.03 (s, 4H); 13C NMR (75 MHz, CDCl3) δ (ppm) 161.7, 147.2, 138.01, 129.8, 129.6, 125.6, 110.0, 29.7; 19F NMR (282 MHz, CDCl3) δ (ppm) −138.11 to −138.29 (m, 4F), −149.90 (t, J = 21.4 Hz, 2F), −157.63 to −157.91 (m, 4F); ESIMS m/z: 717.0 (M + H+, 100%).
General procedure for the nucleophilic aromatic substitution reactions
The reactions of DPP 2 with thiols and phenols were carried out in dry DMF, at room temperature, and in the presence of K2CO3 or Cs2CO3. Once the starting DPP was completely consumed (after 2–3 hours with thiols and 5–6 hours with phenols), the reaction mixtures were diluted with CH2Cl2 and water. The organic layer was then separated and washed with brine and water. The products were isolated by preparative TLC using CH2Cl2/hexane mixtures as the eluent.
Compound 3a. Yield: 51%; mp 274–276 °C; 1H NMR (CDCl3, 300 MHz) δ (ppm) 8.43 (AA’XX’, J = 6 Hz, 4H), 7.68 (AA’BB’, J = 8.7 Hz, 4H), 7.51 (AA’BB’, J = 8.7 Hz, 4H), 6.91 (AA’XX’, J = 6 Hz, 4H), 5.15 (s, 4H); 19F NMR (282 MHz, CDCl3) δ (ppm) −153.53 to −153.65 (m, 4F), −162.60 to −162.73 (m, 4F); ESIMS m/z: 899.1 (M + H+, 100%).
Compound 4a. Yield: 23%; mp 269–273 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 8.45 (AA’XX’, J = 6.3 Hz, 2H), 7.70–7.64 (m, 4H), 7.52 (AA’BB’, J = 8.7 Hz, 4H), 7.07 (AA’XX’, J = 6.3 Hz, 2H), 5.15 (s, 2H), 5.05 (s, 2H); 13C NMR (125 MHz, CDCl3) δ (ppm) 161.7, 149.7, 147.6, 146.95, 145.9, 138.1, 129.9, 129.6, 125.6, 121.1, 109.9, 109.7, 34.9, 34.5; 19F NMR (282 MHz, CDCl3) δ (ppm) −126.79 to −126.90 (m, 2F), –135.53 to –153.81 (m, 2F), −137.99 to −138.50 (m, 2F), −149.74 (t, J = 21.3 Hz, 1F), −157.60 to −157.78 (m, 2H); ESIMS m/z: 802.3 (M + H+, 100%).
Compound 3b. Yield: 85%; mp 270–272 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 8.3–8.32 (m, 2H), 7.68 (AA’BB’, J = 8.7 Hz, 4H), 7.55 (ddd, J = 8.1, 7.4, 1.9 Hz, 2H), 7.49 (AA’BB’, J = 8.7 Hz, 4H), 7.15–7.02 (m, 4H), 5.13 (s, 4H); 13C NMR (125 MHz, CDCl3) δ (ppm) 161.8, 155.5, 150.0, 147.3, 137.9, 137.1, 129.9, 129.5, 125.8, 121.7, 121.1, 116.4, 110.0, 35.1; 19F NMR (282 MHz, CDCl3) δ (ppm) −127.66 to −127.79 (m 4F), −138.03 to −138.25 (m, 4F); ESIMS m/z: 899.0 (M + H+, 100%).
Compound 3c. Yield: 53%; mp 252–256 °C; 1H NMR (300 MHz, DMSO-d6) δ (ppm) 10.08 (s, 2H), 7.78 (AA’BB’, J = 8.7 Hz, 4H), 7.55–7.61 (m, 8H), 7.22 (AA’BB’, J = (8.7 Hz, 4H), 5.09 (s, 4H), 2.03 (s, 6H); 13C NMR (125 MHz, DMSO) δ (ppm) 169.1, 164.7, 161.2, 147.3, 139.9, 136.7, 131.6, 130.9, 130.6, 129.5, 126.4, 125.4, 120.4, 120.1, 109.3, 31.3, 24.5; 19F NMR (282 MHz, DMSO-d6) δ (ppm) −131.31 to −131.54 (m, 4F), −137.94 to −138.07 (m, 4F); ESIMS m/z: 1011.0 (M + H+, 100%).
Compound 3d. Yield: 56%; mp 249–251 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 8.02 (AA’XX’, J = 9 Hz, 4H), 7.68 (AA’BB’, J = 8.7 Hz, 4H), 7.52 (AA’BB’, J = 8.7 Hz, 4H), 6.88 (AA’XX’, J = 9 Hz, 4H), 5.11 (s, 4H), 3.90 (s, 6H); 19F NMR (282 MHz, CDCl3) δ (ppm) −138.23 to −138.35 (m, 4F), −149.59 to −149.96 (m, 4F); ESIMS m/z: 981.0 (M + H+, 100%).
Compound 4d. Yield: 14%; mp 255–257 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 8.02 (AA’XX’, J = 9 Hz, 2H), 7.67–7.59 (m, 4H), 7.52–7.44 (m, 4H), 6.88 (AA’XX’, J = 9 Hz, 2H), 5.10 (s, 2H), 5.03 (s, 2H), 3.92 (s, 3H); 19F NMR (282 MHz, CDCl3) δ (ppm) −138.14 to −138.37 (m, 4F), −149.82 to −150.01 (m, 3F), −157.70 to −157.85 (m, 2F); ESIMS m/z: 849.0 (M + H+, 100%).
Compound 3e. Yield: 63%; mp 262–265 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 7.66 (AA’BB’, J = 8.7 Hz, 4H), 7.47 (AA’BB’, J = 8.7 Hz, 4H), 7.28 (AA’BB’, J = 9 Hz, 4H), 6.75 (AA’BB’, J = 9 Hz, 4H), 5.09 (s, 4H), 1.70 (s, 4H), 1.34 (s, 12H), 0.70 (s, 18H); 13C NMR (125 MHz, DMSO) δ (ppm) 161.7, 154.7, 147.3, 145.8, 137.9, 129.9, 129.4, 127.4, 125.8, 114.8, 109.8, 57.0, 38.2, 34.5, 32.3, 31.8, 31.6; 19F NMR (282 MHz, CDCl3) δ (ppm) −139.07 to −139.30 (m, 4F), −150.36 to −150.46 (m, 4F); ESIMS m/z: 1089.2 (M + H+, 100%).
Compound 3f. Yield: 45%; mp 253–255 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 7.69 (AA’BB’, J = 8.7 Hz, 4H), 7.55 (AA’BB’, J = 8.7 Hz, 4H), 7.25–7.21 (m, 4H), 6.48 (d, J = 8.1 Hz, 4H), 5.12 (s, 4H); 19F NMR (282 MHz, CDCl3) δ (ppm) −137.72 to −137.84 (m, 4F), −145.60 to −145.71 (m, 4F); ESIMS m/z: 867.1 (M + H+, 100%).
Compound 4f. Yield: 13%; mp 248–250 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 7.73–7.58 (m, 4H), 7.58–7.46 (m, 4H), 7.28–7.25 (m, 2H), 6.53 (d, J = 7.8 Hz, 2H), 5.10 (s, 2H), 5.04 (s, 2H); 13C NMR (125 MHz, DMSO) δ (ppm) 177.7, 161.2, 147.4, 141.8, 136.9, 131.1, 130.9, 129.6, 126.3, 118.0, 109.4, 34.9, 34.5; 19F NMR (282 MHz, CDCl3) δ (ppm): −135.56 to −135.69 (m, 2F), −138.23 to −138.34 (m, 2F), −144.36 to −144.49 (m, 2F), −149.67 (t, J = 20.8 Hz, 1F), −157.53 to −157.84 (m, 2F); ESIMS m/z: 792.1 (M + H+, 100%).
Supporting Information
| Supporting Information File 1: 1H NMR, 13C NMR and 19F NMR spectra; MS, UV–vis and emission spectra. | ||
| Format: PDF | Size: 2.6 MB | Download |
Funding
Thanks are due to the University of Aveiro and FCT/MCTES for the financial support for LAQV-REQUIMTE. This work received financial support from FCT/MCTES (Fundação para a Ciência e a Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through projects LA/P/0008/2020 doi:10.54499/LA/P/0008/2020, UIDP/50006/2020 doi:10.54499/UIDP/50006/2020 and UIDB/50006/2020 doi:54499/UIDB/50006/2020, through PT national funds within the PT2020 Partnership Agreement, and to the Portuguese NMR Network. Vítor A. S. Almodovar thanks FCT/MCTES for his doctoral grant (SFRH/BD/135598/2018).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Farnum, D. G.; Mehta, G.; Moore, G. G. I.; Siegal, F. P. Tetrahedron Lett. 1974, 15, 2549–2552. doi:10.1016/s0040-4039(01)93202-2
Return to citation in text: [1] -
Rochat, A. C.; Cassar, L.; Iqbal, A. Preparation of pyrrolo-(3,4-c)-pyrroles. Eur. Pat. Appl. EP0094911A2, Nov 23, 1983.
Return to citation in text: [1] -
Grzybowski, M.; Gryko, D. T. Adv. Opt. Mater. 2015, 3, 280–320. doi:10.1002/adom.201400559
Return to citation in text: [1] [2] -
Głowacki, E. D.; Coskun, H.; Blood-Forsythe, M. A.; Monkowius, U.; Leonat, L.; Grzybowski, M.; Gryko, D.; White, M. S.; Aspuru-Guzik, A.; Sariciftci, N. S. Org. Electron. 2014, 15, 3521–3528. doi:10.1016/j.orgel.2014.09.038
Return to citation in text: [1] -
Mizuguchi, J.; Wooden, G. Ber. Bunsen-Ges. 1991, 95, 1264–1274. doi:10.1002/bbpc.19910951016
Return to citation in text: [1] -
Mizuguchi, J.; Rihs, G. Ber. Bunsen-Ges. 1992, 96, 597–606. doi:10.1002/bbpc.19920960414
Return to citation in text: [1] -
Closs, F.; Gompper, R. Angew. Chem., Int. Ed. Engl. 1987, 26, 552–554. doi:10.1002/anie.198705521
Return to citation in text: [1] -
Liu, Q.; Bottle, S. E.; Sonar, P. Adv. Mater. (Weinheim, Ger.) 2020, 32, 1903882. doi:10.1002/adma.201903882
Return to citation in text: [1] -
Privado, M.; Dahiya, H.; de la Cruz, P.; Keshtov, M. L.; Langa, F.; Sharma, G. D. J. Mater. Chem. C 2021, 9, 16272–16281. doi:10.1039/d1tc02241e
Return to citation in text: [1] -
Zhao, C.; Wang, J.; Jiao, J.; Huang, L.; Tang, J. J. Mater. Chem. C 2020, 8, 28–43. doi:10.1039/c9tc05567c
Return to citation in text: [1] -
Kaur, M.; Choi, D. H. Chem. Soc. Rev. 2015, 44, 58–77. doi:10.1039/c4cs00248b
Return to citation in text: [1] -
Auwalu, M. A.; Cheng, S. Chemosensors 2021, 9, 44. doi:10.3390/chemosensors9030044
Return to citation in text: [1] -
Li, W.; Wang, L.; Tang, H.; Cao, D. Dyes Pigm. 2019, 162, 934–950. doi:10.1016/j.dyepig.2018.11.023
Return to citation in text: [1] -
Schmitt, J.; Heitz, V.; Sour, A.; Bolze, F.; Kessler, P.; Flamigni, L.; Ventura, B.; Bonnet, C. S.; Tóth, É. Chem. – Eur. J. 2016, 22, 2775–2786. doi:10.1002/chem.201503433
Return to citation in text: [1] -
Cai, Y.; Liang, P.; Tang, Q.; Yang, X.; Si, W.; Huang, W.; Zhang, Q.; Dong, X. ACS Nano 2017, 11, 1054–1063. doi:10.1021/acsnano.6b07927
Return to citation in text: [1] -
Agazzi, M. L.; Almodovar, V. A. S.; Gsponer, N. S.; Bertolotti, S.; Tomé, A. C.; Durantini, E. N. Org. Biomol. Chem. 2020, 18, 1449–1461. doi:10.1039/c9ob02487e
Return to citation in text: [1] -
Jiang, X.; Wang, L.; Tang, H.; Cao, D.; Chen, W. Dyes Pigm. 2020, 181, 108599. doi:10.1016/j.dyepig.2020.108599
Return to citation in text: [1] -
Shaikh, S. A. L.; Birajdar, S. S.; Ambore, S. D.; Puyad, A. L.; Vijayanand, P.; Bhosale, S. V.; Bhosale, S. V. Results Chem. 2022, 4, 100473. doi:10.1016/j.rechem.2022.100473
Return to citation in text: [1] -
Cai, Y.; Tang, Q.; Wu, X.; Si, W.; Zhang, Q.; Huang, W.; Dong, X. ACS Appl. Mater. Interfaces 2016, 8, 10737–10742. doi:10.1021/acsami.6b01533
Return to citation in text: [1] -
Cai, Y.; Liang, P.; Tang, Q.; Si, W.; Chen, P.; Zhang, Q.; Dong, X. ACS Appl. Mater. Interfaces 2017, 9, 30398–30405. doi:10.1021/acsami.7b09025
Return to citation in text: [1] -
Zheng, L.; Li, J.; Yu, M.; Jia, W.; Duan, S.; Cao, D.; Ding, X.; Yu, B.; Zhang, X.; Xu, F.-J. J. Am. Chem. Soc. 2020, 142, 20257–20269. doi:10.1021/jacs.0c10771
Return to citation in text: [1] -
Sun, W.; Liu, X.-Y.; Ma, L.-L.; Lu, Z.-L. ACS Appl. Mater. Interfaces 2020, 12, 10193–10201. doi:10.1021/acsami.0c00652
Return to citation in text: [1] -
Gutkowski, K.; Azarias, C.; Banasiewicz, M.; Kozankiewicz, B.; Jacquemin, D.; Gryko, D. T. Eur. J. Org. Chem. 2018, 6643–6648. doi:10.1002/ejoc.201701593
Return to citation in text: [1] -
Jiang, W.; Liu, Z.; Zhu, D.; Zheng, W.; Chen, L.; Zhang, X.; Zhang, G.; Yi, Y.; Jiang, L.; Zhang, D. Angew. Chem., Int. Ed. 2021, 60, 10700–10708. doi:10.1002/anie.202102131
Return to citation in text: [1] -
Vala, M.; Krajčovič, J.; Luňák, S., Jr.; Ouzzane, I.; Bouillon, J.-P.; Weiter, M. Dyes Pigm. 2014, 106, 136–142. doi:10.1016/j.dyepig.2014.03.005
Return to citation in text: [1] -
Beninatto, R.; Borsato, G.; De Lucchi, O.; Fabris, F.; Lucchini, V.; Zendri, E. Dyes Pigm. 2013, 96, 679–685. doi:10.1016/j.dyepig.2012.11.011
Return to citation in text: [1] -
Almodôvar, V. A. S.; Tomé, A. C. Molecules 2021, 26, 4758. doi:10.3390/molecules26164758
Return to citation in text: [1] -
Regeni, I.; Chowdhury, R.; Terlinden, K.; Horiuchi, S.; Holstein, J. J.; Feldmann, S.; Clever, G. H. Angew. Chem., Int. Ed. 2023, 62, e202308288. doi:10.1002/anie.202308288
Return to citation in text: [1] -
Ogumi, K.; Nakagawa, T.; Okada, H.; Matsuo, Y. Org. Electron. 2019, 71, 50–57. doi:10.1016/j.orgel.2019.04.036
Return to citation in text: [1] -
Du, X.; Ma, T.; Ge, T.; Chang, Q.; Liu, X.; Cheng, X. J. Mol. Liq. 2022, 351, 118605. doi:10.1016/j.molliq.2022.118605
Return to citation in text: [1] -
Popli, C.; Patil, Y.; Misra, R. Eur. J. Org. Chem. 2018, 6474–6481. doi:10.1002/ejoc.201801072
Return to citation in text: [1] -
Costa, J. I. T.; Tomé, A. C.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S. J. Porphyrins Phthalocyanines 2011, 15, 1116–1133. doi:10.1142/s1088424611004294
Return to citation in text: [1] -
Brittain, W. D. G.; Coxon, C. R. Chem. – Eur. J. 2022, 28, e202103305. doi:10.1002/chem.202103305
Return to citation in text: [1] -
Kikushima, K.; Koyama, H.; Kodama, K.; Dohi, T. Molecules 2021, 26, 1365. doi:10.3390/molecules26051365
Return to citation in text: [1] -
Bhupathiraju, N. V. S. D. K.; Rizvi, W.; Batteas, J. D.; Drain, C. M. Org. Biomol. Chem. 2016, 14, 389–408. doi:10.1039/c5ob01839k
Return to citation in text: [1] -
Calvo-Castro, J.; Morris, G.; Kennedy, A. R.; McHugh, C. J. Cryst. Growth Des. 2016, 16, 2371–2384. doi:10.1021/acs.cgd.6b00157
Return to citation in text: [1] -
Costa, D. C. S.; Pais, V. F.; Silva, A. M. S.; Cavaleiro, J. A. S.; Pischel, U.; Tomé, J. P. C. Tetrahedron Lett. 2014, 55, 4156–4159. doi:10.1016/j.tetlet.2014.05.108
Return to citation in text: [1] -
Khan, T. K.; Rao, M. R.; Ravikanth, M. Eur. J. Org. Chem. 2010, 2314–2323. doi:10.1002/ejoc.200901460
Return to citation in text: [1] -
Kocak, A.; Kurbanli, S.; Malkondu, S. Synth. Commun. 2007, 37, 3697–3708. doi:10.1080/00397910701569254
Return to citation in text: [1] -
Kuwabara, J.; Yamagata, T.; Kanbara, T. Tetrahedron 2010, 66, 3736–3741. doi:10.1016/j.tet.2010.03.067
Return to citation in text: [1]
| 1. | Farnum, D. G.; Mehta, G.; Moore, G. G. I.; Siegal, F. P. Tetrahedron Lett. 1974, 15, 2549–2552. doi:10.1016/s0040-4039(01)93202-2 |
| 2. | Rochat, A. C.; Cassar, L.; Iqbal, A. Preparation of pyrrolo-(3,4-c)-pyrroles. Eur. Pat. Appl. EP0094911A2, Nov 23, 1983. |
| 9. | Privado, M.; Dahiya, H.; de la Cruz, P.; Keshtov, M. L.; Langa, F.; Sharma, G. D. J. Mater. Chem. C 2021, 9, 16272–16281. doi:10.1039/d1tc02241e |
| 10. | Zhao, C.; Wang, J.; Jiao, J.; Huang, L.; Tang, J. J. Mater. Chem. C 2020, 8, 28–43. doi:10.1039/c9tc05567c |
| 37. | Costa, D. C. S.; Pais, V. F.; Silva, A. M. S.; Cavaleiro, J. A. S.; Pischel, U.; Tomé, J. P. C. Tetrahedron Lett. 2014, 55, 4156–4159. doi:10.1016/j.tetlet.2014.05.108 |
| 38. | Khan, T. K.; Rao, M. R.; Ravikanth, M. Eur. J. Org. Chem. 2010, 2314–2323. doi:10.1002/ejoc.200901460 |
| 39. | Kocak, A.; Kurbanli, S.; Malkondu, S. Synth. Commun. 2007, 37, 3697–3708. doi:10.1080/00397910701569254 |
| 8. | Liu, Q.; Bottle, S. E.; Sonar, P. Adv. Mater. (Weinheim, Ger.) 2020, 32, 1903882. doi:10.1002/adma.201903882 |
| 40. | Kuwabara, J.; Yamagata, T.; Kanbara, T. Tetrahedron 2010, 66, 3736–3741. doi:10.1016/j.tet.2010.03.067 |
| 5. | Mizuguchi, J.; Wooden, G. Ber. Bunsen-Ges. 1991, 95, 1264–1274. doi:10.1002/bbpc.19910951016 |
| 6. | Mizuguchi, J.; Rihs, G. Ber. Bunsen-Ges. 1992, 96, 597–606. doi:10.1002/bbpc.19920960414 |
| 7. | Closs, F.; Gompper, R. Angew. Chem., Int. Ed. Engl. 1987, 26, 552–554. doi:10.1002/anie.198705521 |
| 32. | Costa, J. I. T.; Tomé, A. C.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S. J. Porphyrins Phthalocyanines 2011, 15, 1116–1133. doi:10.1142/s1088424611004294 |
| 33. | Brittain, W. D. G.; Coxon, C. R. Chem. – Eur. J. 2022, 28, e202103305. doi:10.1002/chem.202103305 |
| 34. | Kikushima, K.; Koyama, H.; Kodama, K.; Dohi, T. Molecules 2021, 26, 1365. doi:10.3390/molecules26051365 |
| 35. | Bhupathiraju, N. V. S. D. K.; Rizvi, W.; Batteas, J. D.; Drain, C. M. Org. Biomol. Chem. 2016, 14, 389–408. doi:10.1039/c5ob01839k |
| 3. | Grzybowski, M.; Gryko, D. T. Adv. Opt. Mater. 2015, 3, 280–320. doi:10.1002/adom.201400559 |
| 4. | Głowacki, E. D.; Coskun, H.; Blood-Forsythe, M. A.; Monkowius, U.; Leonat, L.; Grzybowski, M.; Gryko, D.; White, M. S.; Aspuru-Guzik, A.; Sariciftci, N. S. Org. Electron. 2014, 15, 3521–3528. doi:10.1016/j.orgel.2014.09.038 |
| 36. | Calvo-Castro, J.; Morris, G.; Kennedy, A. R.; McHugh, C. J. Cryst. Growth Des. 2016, 16, 2371–2384. doi:10.1021/acs.cgd.6b00157 |
| 19. | Cai, Y.; Tang, Q.; Wu, X.; Si, W.; Zhang, Q.; Huang, W.; Dong, X. ACS Appl. Mater. Interfaces 2016, 8, 10737–10742. doi:10.1021/acsami.6b01533 |
| 20. | Cai, Y.; Liang, P.; Tang, Q.; Si, W.; Chen, P.; Zhang, Q.; Dong, X. ACS Appl. Mater. Interfaces 2017, 9, 30398–30405. doi:10.1021/acsami.7b09025 |
| 21. | Zheng, L.; Li, J.; Yu, M.; Jia, W.; Duan, S.; Cao, D.; Ding, X.; Yu, B.; Zhang, X.; Xu, F.-J. J. Am. Chem. Soc. 2020, 142, 20257–20269. doi:10.1021/jacs.0c10771 |
| 22. | Sun, W.; Liu, X.-Y.; Ma, L.-L.; Lu, Z.-L. ACS Appl. Mater. Interfaces 2020, 12, 10193–10201. doi:10.1021/acsami.0c00652 |
| 26. | Beninatto, R.; Borsato, G.; De Lucchi, O.; Fabris, F.; Lucchini, V.; Zendri, E. Dyes Pigm. 2013, 96, 679–685. doi:10.1016/j.dyepig.2012.11.011 |
| 27. | Almodôvar, V. A. S.; Tomé, A. C. Molecules 2021, 26, 4758. doi:10.3390/molecules26164758 |
| 28. | Regeni, I.; Chowdhury, R.; Terlinden, K.; Horiuchi, S.; Holstein, J. J.; Feldmann, S.; Clever, G. H. Angew. Chem., Int. Ed. 2023, 62, e202308288. doi:10.1002/anie.202308288 |
| 3. | Grzybowski, M.; Gryko, D. T. Adv. Opt. Mater. 2015, 3, 280–320. doi:10.1002/adom.201400559 |
| 18. | Shaikh, S. A. L.; Birajdar, S. S.; Ambore, S. D.; Puyad, A. L.; Vijayanand, P.; Bhosale, S. V.; Bhosale, S. V. Results Chem. 2022, 4, 100473. doi:10.1016/j.rechem.2022.100473 |
| 29. | Ogumi, K.; Nakagawa, T.; Okada, H.; Matsuo, Y. Org. Electron. 2019, 71, 50–57. doi:10.1016/j.orgel.2019.04.036 |
| 30. | Du, X.; Ma, T.; Ge, T.; Chang, Q.; Liu, X.; Cheng, X. J. Mol. Liq. 2022, 351, 118605. doi:10.1016/j.molliq.2022.118605 |
| 31. | Popli, C.; Patil, Y.; Misra, R. Eur. J. Org. Chem. 2018, 6474–6481. doi:10.1002/ejoc.201801072 |
| 14. | Schmitt, J.; Heitz, V.; Sour, A.; Bolze, F.; Kessler, P.; Flamigni, L.; Ventura, B.; Bonnet, C. S.; Tóth, É. Chem. – Eur. J. 2016, 22, 2775–2786. doi:10.1002/chem.201503433 |
| 15. | Cai, Y.; Liang, P.; Tang, Q.; Yang, X.; Si, W.; Huang, W.; Zhang, Q.; Dong, X. ACS Nano 2017, 11, 1054–1063. doi:10.1021/acsnano.6b07927 |
| 16. | Agazzi, M. L.; Almodovar, V. A. S.; Gsponer, N. S.; Bertolotti, S.; Tomé, A. C.; Durantini, E. N. Org. Biomol. Chem. 2020, 18, 1449–1461. doi:10.1039/c9ob02487e |
| 17. | Jiang, X.; Wang, L.; Tang, H.; Cao, D.; Chen, W. Dyes Pigm. 2020, 181, 108599. doi:10.1016/j.dyepig.2020.108599 |
| 11. | Kaur, M.; Choi, D. H. Chem. Soc. Rev. 2015, 44, 58–77. doi:10.1039/c4cs00248b |
| 12. | Auwalu, M. A.; Cheng, S. Chemosensors 2021, 9, 44. doi:10.3390/chemosensors9030044 |
| 13. | Li, W.; Wang, L.; Tang, H.; Cao, D. Dyes Pigm. 2019, 162, 934–950. doi:10.1016/j.dyepig.2018.11.023 |
| 23. | Gutkowski, K.; Azarias, C.; Banasiewicz, M.; Kozankiewicz, B.; Jacquemin, D.; Gryko, D. T. Eur. J. Org. Chem. 2018, 6643–6648. doi:10.1002/ejoc.201701593 |
| 24. | Jiang, W.; Liu, Z.; Zhu, D.; Zheng, W.; Chen, L.; Zhang, X.; Zhang, G.; Yi, Y.; Jiang, L.; Zhang, D. Angew. Chem., Int. Ed. 2021, 60, 10700–10708. doi:10.1002/anie.202102131 |
| 25. | Vala, M.; Krajčovič, J.; Luňák, S., Jr.; Ouzzane, I.; Bouillon, J.-P.; Weiter, M. Dyes Pigm. 2014, 106, 136–142. doi:10.1016/j.dyepig.2014.03.005 |
© 2024 Almodovar and Tomé; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.