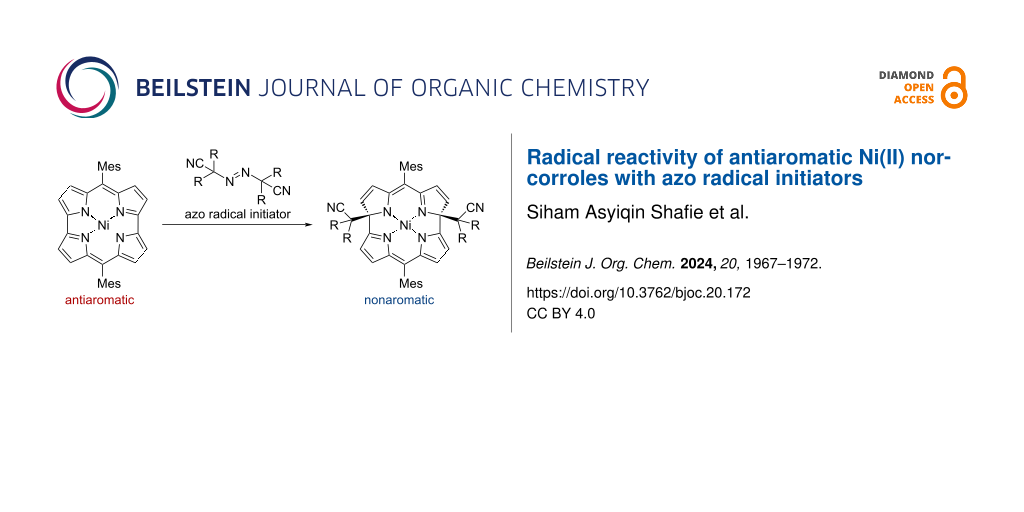
Abstract
Norcorrole is a stable 16π-antiaromatic porphyrinoid that exhibits characteristic reactivities and physical properties. Here, we disclose the reaction of Ni(II) norcorroles with alkyl radicals derived from azo radical initiators. The radical selectively attacked the distal α-position relative to the meso-position to construct a nonaromatic bowl-shaped structure. The photophysical and electrochemical properties of the obtained radical adducts were compared to those of the parent Ni(II) norcorrole. The radical reactivity of Ni(II) norcorroles was investigated by density functional theory (DFT) calculations.

Graphical Abstract
Introduction
Considerable attention has been directed toward antiaromatic norcorroles [1-3] due to the fascinating physical properties, such as reversible redox properties [4,5] and stacked-ring aromaticity [6-10]. While Ni(II) norcorroles are stable under ambient conditions despite the distinct 16π-antiaromaticity, they show unique reactivities with various reagents due to the high-lying HOMO and low-lying LUMO (Figure 1) [11]. Reactions with nucleophiles (Nu) proceed with perfect regioselectivity at the distal β-position relative to the meso-position [12-15]. On the other hand, reactions with electrophiles (El) also occur preferentially at the β-positions, but the regioselectivity depends on the electrophile [16-18]. In addition, C–C double bonds of the norcorrole skeleton outside the π-delocalization pathway exhibit a reactivity similar to an alkene to afford hydrogenated norcorroles by hydrogenation [19] or reduction with hydrazine [20] and [3 + 2]-cycloadducts with 1,3-dipoles [21]. Moreover, the ring-expansion or ring-opening reactions of Ni(II) norcorroles are induced by an activated zwitterionic intermediate [22], oxidants [23,24], and carbenes [25,26].
![[1860-5397-20-172-1]](/bjoc/content/figures/1860-5397-20-172-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Reactivities of norcorroles with various reagents.
Figure 1: Reactivities of norcorroles with various reagents.
During the last decade, the various reactivities of Ni(II) norcorroles have been elucidated. However, the reaction with radical species has remained unexplored. Here, we disclose the radical functionalization of Ni(II) norcorroles with simple and frequently used azo radical initiators to furnish nonconjugated macrocycles with bowl-shaped structures [27]. The photophysical and electronic properties of the obtained products are also presented. We also discuss the selectivity of the radical addition to Ni(II) norcorroles using DFT calculations.
Results and Discussion
Reactivity with azo radical initiators
We selected 2,2'-azobis(isobutyronitrile) (AIBN) as a radical source. Ni(II) dimesitylnorcorrole 1 was treated with AIBN in refluxing toluene (Scheme 1). The reaction smoothly proceeded to afford dialkylated macrocycle 2a in 92% yield. In addition to 2a, monoalkylated product 3a and dipyrrin dimer 4a were obtained as minor products in 4% and 3% yield, respectively.
![[1860-5397-20-172-i1]](/bjoc/content/inline/1860-5397-20-172-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of norcorrole 1 with AIBN.
Scheme 1: Reaction of norcorrole 1 with AIBN.
The structure of 2a was unambiguously confirmed by single-crystal X-ray analysis, which revealed that two alkyl substituents were located on the same side of the molecule (Figure 2a). Compared to the planar structure of 1 (Figure 2b) [2], 2a displays a nonplanar structure due to the sp3 carbon atoms adjacent to the nitrogen atoms. The 1H NMR spectrum of 2a confirmed that the antiaromatic character of the macrocycle changed to nonaromatic upon radical addition (see Supporting Information File 1).
![[1860-5397-20-172-2]](/bjoc/content/figures/1860-5397-20-172-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Top and side views of the X-ray structures of a) 2a and b) 1 [2]. Mesityl groups and hydrogen atoms were omitted for clarity. Thermal ellipsoids are drawn at 50% probability.
Figure 2: Top and side views of the X-ray structures of a) 2a and b) 1 [2]. Mesityl groups and hydrogen atoms we...
1,1'-Azobis(cyclohexane-1-carbonitrile) (V-40) was also examined as a radical source. The reaction afforded 2b in 87% yield (Scheme 2). Unfortunately, other radical sources, such as benzoyl peroxide, TEMPO, and the combination of alkyl halides with BEt3, were not applicable to this reaction.
![[1860-5397-20-172-i2]](/bjoc/content/inline/1860-5397-20-172-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of norcorrole 1 with V-40.
Scheme 2: Reaction of norcorrole 1 with V-40.
Physical properties
The electronic absorption spectra of norcorrole 1 and adduct 2a are shown in Figure 3. While norcorrole 1 exhibited a weak absorption band from 600 nm to the NIR region, due to the characteristic forbidden HOMO–LUMO transition of the antiaromatic compound, nonconjugated macrocycle 2a did not possess such an absorption band, indicating the loss of antiaromaticity in 2a. Macrocycle 2a possessed new absorption bands from 600 to 800 nm. The simulated absorption spectrum of 2a obtained by TD DFT calculations at the M06/6-31G(d)+SDD//B3LYP-D3/6-31G(d)+SDD level of theory was consistent with the experimental results. Therein, the absorption band at 670 nm (f = 0.0026) was attributed to the transition from HOMO to LUMO+1.
![[1860-5397-20-172-3]](/bjoc/content/figures/1860-5397-20-172-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis–NIR absorption spectra of 1 and 2a in CH2Cl2.
Figure 3: UV–vis–NIR absorption spectra of 1 and 2a in CH2Cl2.
Next, the electrochemical properties of 2a in CH2Cl2 were examined using cyclic voltammetry (Figure 4). Macrocycle 2a exhibited one reversible oxidation wave at 0.44 V and two reversible reduction waves at −0.85 V and −1.14 V. The electrochemical HOMO–LUMO gap of 2a is 1.29 V, which is larger than that of 1a (1.08 V) [2].
![[1860-5397-20-172-4]](/bjoc/content/figures/1860-5397-20-172-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Cyclic voltammogram of 2a in CH2Cl2. Supporting electrolyte: 0.1 M Bu4NPF6; working electrode: glassy carbon; counter electrode: Pt; reference electrode: Ag/AgNO3; scan rate: 50 mV⋅s−1.
Figure 4: Cyclic voltammogram of 2a in CH2Cl2. Supporting electrolyte: 0.1 M Bu4NPF6; working electrode: glas...
DFT calculations
We next conducted DFT calculations using Gaussian 16 [28] to elucidate the reactivity of Ni(II) norcorroles with radical species (Scheme 3). All calculations for the ground state were performed at the (U)B3LYP-D3/6-31G(d)+SDD level of theory. The SOMO of an isobutyronitrile radical (−5.98 eV), which was generated through denitrogenation of AIBN, is closer to the HOMO level of Ni(II) norcorrole 1 (−4.68 eV) rather than its LUMO (−3.16 eV). This result explains the selective addition of the electrophilic isobutyronitrile radical to the distal α-position of the pyrrole unit. The calculated molecular orbital coefficient of the HOMO indicates that two α-carbon atoms of the pyrrole subunits are the most reactive positions for electrophilic species. In addition, the distal α-carbon atom relative to the meso-position could be more reactive than the proximal α-carbon atom due to the steric hindrance of bulky mesityl groups. Consequently, the isobutyronitrile radical predominantly attacks the distal α-carbon atom relative to the meso-position to afford the corresponding radical intermediate I. The calculated spin density of radical I revealed a substantial radical character at the α-position of the pyrrole skeleton. Finally, another isobutyronitrile radical reacts with I at the convex face to form the major product 2a, with two alkyl substituents on the same side of the molecule. The mean-plane deviation (MPD) of I was 0.293 Å, where the mean plane was defined by carbon, nitrogen, and nickel atoms of the norcorrole core. For the byproducts, 3a would be generated through the quenching of radical I with a hydrogen atom source. Bisdipyrrin 4a could be formed through the ring-opening reaction of I by the homolytic cleavage of the C(sp2)–C(sp2) bond to radical II, the addition of the isobutyronitrile radical, and subsequent demetallation.
![[1860-5397-20-172-i3]](/bjoc/content/inline/1860-5397-20-172-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have investigated the addition reaction of electrophilic alkyl radicals derived from azo radical initiators to antiaromatic Ni(II) norcorroles. The reaction smoothly proceeded to afford bowl-shaped nonconjugated macrocycles 2a in excellent yield, which exhibited markedly different photophysical and electrochemical properties with norcorrole 1. The intrinsic reactivities of Ni(II) norcorroles with neutral radical species were revealed by DFT calculations, where populations of the HOMO of the norcorrole unit and the spin density of the radical intermediate governed the regioselectivity.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data including NMR and MS spectra, additional crystal data and details from DFT calculations. | ||
| Format: PDF | Size: 1.1 MB | Download |
Funding
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grants JP20H05863, JP22H04974, and JP22K19025. H. T. is grateful to the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Leading Initiative for Excellent Young Researchers (Grant JPMXS0320220200) and the Foundation of Public Interest Tatematsu.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Bröring, M.; Köhler, S.; Kleeberg, C. Angew. Chem., Int. Ed. 2008, 47, 5658–5660. doi:10.1002/anie.200801196
Return to citation in text: [1] -
Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.-Y.; Kobayashi, N.; Shinokubo, H. Angew. Chem., Int. Ed. 2012, 51, 8542–8545. doi:10.1002/anie.201204395
Return to citation in text: [1] [2] [3] [4] -
Yonezawa, T.; Shafie, S. A.; Hiroto, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2017, 56, 11822–11825. doi:10.1002/anie.201706134
Return to citation in text: [1] -
Shin, J.-Y.; Yamada, T.; Yoshikawa, H.; Awaga, K.; Shinokubo, H. Angew. Chem., Int. Ed. 2014, 53, 3096–3101. doi:10.1002/anie.201310374
Return to citation in text: [1] -
Ukai, S.; Fukui, N.; Ikeue, T.; Shinokubo, H. Chem. Lett. 2022, 51, 182–184. doi:10.1246/cl.210715
Return to citation in text: [1] -
Nozawa, R.; Tanaka, H.; Cha, W.-Y.; Hong, Y.; Hisaki, I.; Shimizu, S.; Shin, J.-Y.; Kowalczyk, T.; Irle, S.; Kim, D.; Shinokubo, H. Nat. Commun. 2016, 7, 13620. doi:10.1038/ncomms13620
Return to citation in text: [1] -
Kawashima, H.; Ukai, S.; Nozawa, R.; Fukui, N.; Fitzsimmons, G.; Kowalczyk, T.; Fliegl, H.; Shinokubo, H. J. Am. Chem. Soc. 2021, 143, 10676–10685. doi:10.1021/jacs.1c04348
Return to citation in text: [1] -
Kawashima, H.; Fukui, N.; Phung, Q. M.; Yanai, T.; Shinokubo, H. Cell Rep. Phys. Sci. 2022, 3, 101045. doi:10.1016/j.xcrp.2022.101045
Return to citation in text: [1] -
Ishikawa, S.; Yamasumi, K.; Sugiura, S.; Sato, S.; Watanabe, G.; Koo, Y. H.; Seki, S.; Bando, Y.; Haketa, Y.; Shinokubo, H.; Maeda, H. Chem. Sci. 2024, 15, 7603–7609. doi:10.1039/d4sc01633e
Return to citation in text: [1] -
Kino, S.; Ukai, S.; Fukui, N.; Haruki, R.; Kumai, R.; Wang, Q.; Horike, S.; Phung, Q. M.; Sundholm, D.; Shinokubo, H. J. Am. Chem. Soc. 2024, 146, 9311–9317. doi:10.1021/jacs.4c01142
Return to citation in text: [1] -
Li, S.; Sun, Y.; Meng, Y.; Li, X.; Zhang, S. Chin. J. Org. Chem. 2022, 42, 2390. doi:10.6023/cjoc202202039
Return to citation in text: [1] -
Nozawa, R.; Yamamoto, K.; Shin, J.-Y.; Hiroto, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2015, 54, 8454–8457. doi:10.1002/anie.201502666
Return to citation in text: [1] -
Liu, B.; Yoshida, T.; Li, X.; Stępień, M.; Shinokubo, H.; Chmielewski, P. J. Angew. Chem., Int. Ed. 2016, 55, 13142–13146. doi:10.1002/anie.201607237
Return to citation in text: [1] -
Yoshida, T.; Shinokubo, H. Mater. Chem. Front. 2017, 1, 1853–1857. doi:10.1039/c7qm00176b
Return to citation in text: [1] -
Ren, D.; Fu, X.; Li, X.; Koniarz, S.; Chmielewski, P. J. Org. Chem. Front. 2019, 6, 2924–2933. doi:10.1039/c9qo00679f
Return to citation in text: [1] -
Deng, Z.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. – Eur. J. 2016, 22, 4231–4246. doi:10.1002/chem.201504584
Return to citation in text: [1] -
Kawashima, H.; Hiroto, S.; Shinokubo, H. J. Org. Chem. 2017, 82, 10425–10432. doi:10.1021/acs.joc.7b01899
Return to citation in text: [1] -
Li, S.; Smaga, O.; Sun, Y.; Li, X.; Pawlicki, M.; Sukniewicz, M.; Chmielewski, P. J. Org. Chem. Front. 2021, 8, 3639–3652. doi:10.1039/d1qo00621e
Return to citation in text: [1] -
Liu, B.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. – Eur. J. 2015, 21, 7790–7797. doi:10.1002/chem.201500736
Return to citation in text: [1] -
Nozawa, R.; Yamamoto, K.; Hisaki, I.; Shin, J.-Y.; Shinokubo, H. Chem. Commun. 2016, 52, 7106–7109. doi:10.1039/c6cc02918c
Return to citation in text: [1] -
Fu, X.; Meng, Y.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. Commun. 2018, 54, 2510–2513. doi:10.1039/c8cc00447a
Return to citation in text: [1] -
Ren, D.; Smaga, O.; Fu, X.; Li, X.; Pawlicki, M.; Koniarz, S.; Chmielewski, P. J. Org. Lett. 2021, 23, 1032–1037. doi:10.1021/acs.orglett.0c04227
Return to citation in text: [1] -
Liu, S.-Y.; Tanaka, H.; Nozawa, R.; Fukui, N.; Shinokubo, H. Chem. – Eur. J. 2019, 25, 7618–7622. doi:10.1002/chem.201901292
Return to citation in text: [1] -
Shafie, S. A.; Kawashima, H.; Miyake, Y.; Shinokubo, H. ChemPlusChem 2019, 84, 623–626. doi:10.1002/cplu.201900068
Return to citation in text: [1] -
Fukuoka, T.; Uchida, K.; Sung, Y. M.; Shin, J.-Y.; Ishida, S.; Lim, J. M.; Hiroto, S.; Furukawa, K.; Kim, D.; Iwamoto, T.; Shinokubo, H. Angew. Chem., Int. Ed. 2014, 53, 1506–1509. doi:10.1002/anie.201309921
Return to citation in text: [1] -
Liu, S.-Y.; Fukuoka, T.; Fukui, N.; Shin, J.-Y.; Shinokubo, H. Org. Lett. 2020, 22, 4400–4403. doi:10.1021/acs.orglett.0c01402
Return to citation in text: [1] -
Tabata, N.; Uchino, T.; Kitamura, C.; Yoshizawa, K.; Shiota, Y.; Kato, S.-i. Chem. Sci. 2023, 14, 5974–5982. doi:10.1039/d3sc00381g
Return to citation in text: [1] -
Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1]
| 1. | Bröring, M.; Köhler, S.; Kleeberg, C. Angew. Chem., Int. Ed. 2008, 47, 5658–5660. doi:10.1002/anie.200801196 |
| 2. | Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.-Y.; Kobayashi, N.; Shinokubo, H. Angew. Chem., Int. Ed. 2012, 51, 8542–8545. doi:10.1002/anie.201204395 |
| 3. | Yonezawa, T.; Shafie, S. A.; Hiroto, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2017, 56, 11822–11825. doi:10.1002/anie.201706134 |
| 12. | Nozawa, R.; Yamamoto, K.; Shin, J.-Y.; Hiroto, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2015, 54, 8454–8457. doi:10.1002/anie.201502666 |
| 13. | Liu, B.; Yoshida, T.; Li, X.; Stępień, M.; Shinokubo, H.; Chmielewski, P. J. Angew. Chem., Int. Ed. 2016, 55, 13142–13146. doi:10.1002/anie.201607237 |
| 14. | Yoshida, T.; Shinokubo, H. Mater. Chem. Front. 2017, 1, 1853–1857. doi:10.1039/c7qm00176b |
| 15. | Ren, D.; Fu, X.; Li, X.; Koniarz, S.; Chmielewski, P. J. Org. Chem. Front. 2019, 6, 2924–2933. doi:10.1039/c9qo00679f |
| 2. | Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.-Y.; Kobayashi, N.; Shinokubo, H. Angew. Chem., Int. Ed. 2012, 51, 8542–8545. doi:10.1002/anie.201204395 |
| 11. | Li, S.; Sun, Y.; Meng, Y.; Li, X.; Zhang, S. Chin. J. Org. Chem. 2022, 42, 2390. doi:10.6023/cjoc202202039 |
| 2. | Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.-Y.; Kobayashi, N.; Shinokubo, H. Angew. Chem., Int. Ed. 2012, 51, 8542–8545. doi:10.1002/anie.201204395 |
| 6. | Nozawa, R.; Tanaka, H.; Cha, W.-Y.; Hong, Y.; Hisaki, I.; Shimizu, S.; Shin, J.-Y.; Kowalczyk, T.; Irle, S.; Kim, D.; Shinokubo, H. Nat. Commun. 2016, 7, 13620. doi:10.1038/ncomms13620 |
| 7. | Kawashima, H.; Ukai, S.; Nozawa, R.; Fukui, N.; Fitzsimmons, G.; Kowalczyk, T.; Fliegl, H.; Shinokubo, H. J. Am. Chem. Soc. 2021, 143, 10676–10685. doi:10.1021/jacs.1c04348 |
| 8. | Kawashima, H.; Fukui, N.; Phung, Q. M.; Yanai, T.; Shinokubo, H. Cell Rep. Phys. Sci. 2022, 3, 101045. doi:10.1016/j.xcrp.2022.101045 |
| 9. | Ishikawa, S.; Yamasumi, K.; Sugiura, S.; Sato, S.; Watanabe, G.; Koo, Y. H.; Seki, S.; Bando, Y.; Haketa, Y.; Shinokubo, H.; Maeda, H. Chem. Sci. 2024, 15, 7603–7609. doi:10.1039/d4sc01633e |
| 10. | Kino, S.; Ukai, S.; Fukui, N.; Haruki, R.; Kumai, R.; Wang, Q.; Horike, S.; Phung, Q. M.; Sundholm, D.; Shinokubo, H. J. Am. Chem. Soc. 2024, 146, 9311–9317. doi:10.1021/jacs.4c01142 |
| 27. | Tabata, N.; Uchino, T.; Kitamura, C.; Yoshizawa, K.; Shiota, Y.; Kato, S.-i. Chem. Sci. 2023, 14, 5974–5982. doi:10.1039/d3sc00381g |
| 4. | Shin, J.-Y.; Yamada, T.; Yoshikawa, H.; Awaga, K.; Shinokubo, H. Angew. Chem., Int. Ed. 2014, 53, 3096–3101. doi:10.1002/anie.201310374 |
| 5. | Ukai, S.; Fukui, N.; Ikeue, T.; Shinokubo, H. Chem. Lett. 2022, 51, 182–184. doi:10.1246/cl.210715 |
| 2. | Ito, T.; Hayashi, Y.; Shimizu, S.; Shin, J.-Y.; Kobayashi, N.; Shinokubo, H. Angew. Chem., Int. Ed. 2012, 51, 8542–8545. doi:10.1002/anie.201204395 |
| 21. | Fu, X.; Meng, Y.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. Commun. 2018, 54, 2510–2513. doi:10.1039/c8cc00447a |
| 23. | Liu, S.-Y.; Tanaka, H.; Nozawa, R.; Fukui, N.; Shinokubo, H. Chem. – Eur. J. 2019, 25, 7618–7622. doi:10.1002/chem.201901292 |
| 24. | Shafie, S. A.; Kawashima, H.; Miyake, Y.; Shinokubo, H. ChemPlusChem 2019, 84, 623–626. doi:10.1002/cplu.201900068 |
| 20. | Nozawa, R.; Yamamoto, K.; Hisaki, I.; Shin, J.-Y.; Shinokubo, H. Chem. Commun. 2016, 52, 7106–7109. doi:10.1039/c6cc02918c |
| 25. | Fukuoka, T.; Uchida, K.; Sung, Y. M.; Shin, J.-Y.; Ishida, S.; Lim, J. M.; Hiroto, S.; Furukawa, K.; Kim, D.; Iwamoto, T.; Shinokubo, H. Angew. Chem., Int. Ed. 2014, 53, 1506–1509. doi:10.1002/anie.201309921 |
| 26. | Liu, S.-Y.; Fukuoka, T.; Fukui, N.; Shin, J.-Y.; Shinokubo, H. Org. Lett. 2020, 22, 4400–4403. doi:10.1021/acs.orglett.0c01402 |
| 19. | Liu, B.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. – Eur. J. 2015, 21, 7790–7797. doi:10.1002/chem.201500736 |
| 16. | Deng, Z.; Li, X.; Stępień, M.; Chmielewski, P. J. Chem. – Eur. J. 2016, 22, 4231–4246. doi:10.1002/chem.201504584 |
| 17. | Kawashima, H.; Hiroto, S.; Shinokubo, H. J. Org. Chem. 2017, 82, 10425–10432. doi:10.1021/acs.joc.7b01899 |
| 18. | Li, S.; Smaga, O.; Sun, Y.; Li, X.; Pawlicki, M.; Sukniewicz, M.; Chmielewski, P. J. Org. Chem. Front. 2021, 8, 3639–3652. doi:10.1039/d1qo00621e |
| 22. | Ren, D.; Smaga, O.; Fu, X.; Li, X.; Pawlicki, M.; Koniarz, S.; Chmielewski, P. J. Org. Lett. 2021, 23, 1032–1037. doi:10.1021/acs.orglett.0c04227 |
© 2024 Shafie et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.