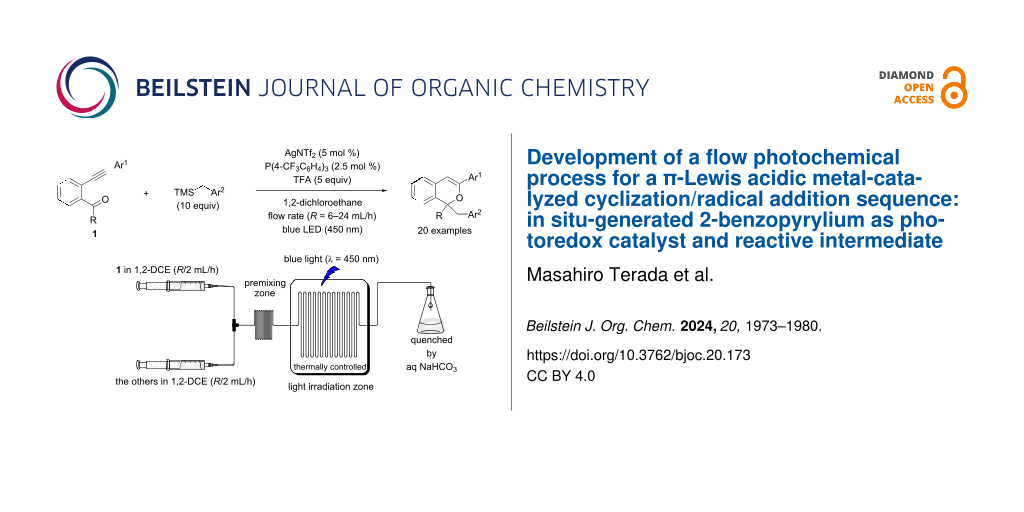
Abstract
A flow photochemical reaction system for a π-Lewis acidic metal-catalyzed cyclization/radical addition sequence was developed, which utilizes in situ-generated 2-benzopyrylium intermediates as the photoredox catalyst and electrophilic substrates. The key 2-benzopyrylium intermediates were generated in the flow reaction system through the intramolecular cyclization of ortho-carbonyl alkynylbenzene derivatives by the π-Lewis acidic metal catalyst AgNTf2 and the subsequent proto-demetalation with trifluoroacetic acid. The 2-benzopyrylium intermediates underwent further photoreactions with benzyltrimethylsilane derivatives as the donor molecule in the flow photoreactor to provide 1H-isochromene derivatives in higher yields in most cases than the batch reaction system.

Graphical Abstract
Introduction
Flow chemistry has been actively studied in recent years as a method to run a reaction continuously using a flow path or tube, rather than in a flask [1-16]. This method has attracted much attention because, unlike a batch reaction system, it allows for rapid generation of unstable chemical species by controlling parameters such as flow velocity and mixing properties, and in some cases makes it possible to achieve reactions that are difficult to perform using batch chemistry [17-21]. In general, efficient two-phase mixing and heat transfer, as well as ease of scale-up, are the advantages of using a flow system. In addition, reproducibility in a liquid–liquid flow system is improved because the flow velocity and temperature can be precisely controlled by using a syringe pump and a temperature control unit, respectively. Moreover, as the reaction mixture continues to flow and the reaction can be quenched immediately when necessary, the decomposition of an unstable product under the reaction conditions can be avoided [22-25]. Furthermore, when a photoreaction is performed in a flow system, there is an advantage that the light irradiation efficiency [26-29] is increased. Thus, the flow photochemical process is crucial and beneficial to product formation.
Recently, we reported a sequential transformation consisting of a π-Lewis acidic metal-catalyzed cyclization [30-45] and subsequent photochemical radical addition [46-54], which affords 1H-isochromene derivatives 3 through three catalytic cycles (Scheme 1a) [55]: catalytic cycles I and II and a photoredox cycle of the photocatalyst [56,57] (see Supporting Information File 1 for the overall catalytic cycles). In catalytic cycle I, the key cationic components, 2-benzopyrylium intermediates A, are generated in situ by the activation of the alkyne moiety of ortho-carbonyl alkynylbenzene derivatives 1 in the presence of the π-Lewis acidic metal catalyst [M]X [AgNTf2 or Cu(NTf2)2] and subsequent intramolecular cyclization followed by proto-demetalation with trifluoroacetic acid (TFA). In catalytic cycle II, photoexcitation of the generated 2-benzopyrylium intermediates A under light irradiation facilitates single-electron transfer (SET) from benzyltrimethylsilane derivatives 2 as the donor molecule, initiating further radical reactions through the formation of radical cations B. Nucleophilic arylmethyl radicals C, which are generated from radical cations B by desilylation, undergo an addition reaction with 2-benzopyrylium intermediates A, giving rise to the corresponding radical cation. Catalytic cycle II is completed through a SET from D, a reduced form of the photoredox catalyst 2-benzopyrylium intermediates A, to the generated radical cation, affording 1H-isochromene derivatives 3. The photoredox cycle is also completed with the regeneration of cations A through SET from D.
![[1860-5397-20-173-i1]](/bjoc/content/inline/1860-5397-20-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) Sequential π-Lewis acidic metal-catalyzed cyclization/photochemical radical addition for the formation of 1H-isochromene derivatives 3 and its plausible catalytic cycles. (b) Stability of 3a under the optimal reaction conditions of the batch reaction.
Scheme 1: (a) Sequential π-Lewis acidic metal-catalyzed cyclization/photochemical radical addition for the fo...
The most distinctive feature of this sequential transformation is that the in situ-generated 2-benzopyrylium intermediates A are used not only as an electrophile but also as a photoredox catalyst. However, as this reaction is carried out under relatively harsh conditions (i.e., light irradiation, use of an excess amount of TFA), the stability of products 3 was a concern. Indeed, subjecting product 3a to the optimal reaction conditions with either AgNTf2 or Cu(NTf2)2 resulted in the significant degradation of 3a, although the degradation of 3a was partially suppressed when AgNTf2 was used (Scheme 1b). Accordingly, we envisioned that the characteristics of the flow photochemical process, i.e., efficient light irradiation and immediate separation of the formed product from the reaction system, would be suitable for this sequential reaction. Here, we report the results of our investigation on the use of a flow photochemical reaction system to improve the yield of the present sequential transformation.
Results and Discussion
At the outset of our studies to optimize the flow reaction conditions, we employed AgNTf2 as the π-Lewis acidic metal catalyst because of its high solubility in 1,2-dichloroethane (1,2-DCE) [58] and ability to partially suppress the degradation of the product formed (Scheme 1b). When designing a flow reaction system for the present sequential transformation, we considered the fact that the transformation involves three catalytic cycles. In particular, given that catalytic cycle I (see Scheme 1a) generates, e.g., key cationic components, 2-benzopyrylium intermediates A without light irradiation, it is necessary to ensure that the reaction time of catalytic cycle I is not affected by the timescale of the flow reaction. Therefore, we adopted a dual syringe system in which two solutions are mixed before being introduced into the photoreactor (volume: 1.0 mL) (Table 1, top right). After several trials, we decided to fill syringe A with o-alkynylacetophenone 1a and syringe B with AgNTf2, P(4-CF3C6H4)3, benzyltrimethylsilane (2a, TMSBn), and TFA (see Supporting Information File 1 for details). At this time, the volumes of the solutions placed in the two syringes were adjusted to be approximately the same. The initial conditions of the flow reaction were based on those of the batch reaction [0.1 mmol of 1a, 10 μmol (10 mol %) of AgNTf2, 20 μmol (20 mol %) of P(4-CF3C6H4)3, 1.0 mmol (10 equiv) of 2a, and 0.5 mmol (5 equiv) of TFA under light irradiation (blue LED: λmax = 448 nm) at 50 °C for 1 h in 1 mL (total volume) of 1,2-DCE] [55] with a flow rate of 3 mL/h (light irradiation time: 20 min in the flow reaction, 1 h in the batch reaction). As shown in Table 1, product 3a was obtained in moderate yield (entry 1: 42%, cf. batch reaction: 76%). Lowering the reaction temperature to 25 °C reduced the yield (Table 1, entry 2: 35%), but decreasing the amount of the phosphine ligand from 20 mol % to 5 mol % markedly improved the yield (Table 1, entry 3: 53%). Even when the flow rate was increased from 3 mL/h to 24 mL/h (light irradiation time was shortened from 20 min to 2.5 min), the yield of 3a was maintained (Table 1, entry 4: 53%). Under these conditions, no improvement in yield was observed when the premixing zone (0.7 mL) was provided (Table 1, entry 5: 52%); however, the effect of adding the premixing zone was remarkable when the amount of AgNTf2 was reduced by half (5 mol %; Table 1, entry 6 vs entry 7: 26% vs 49%). These results suggest that the generation of 2-benzopyrylium intermediates A, (i.e., catalytic cycle I) requires a certain reaction time (at this flow rate: ca. 2 min). Moreover, the yield of 3a was improved when the concentration of 1a was lowered from 0.1 M to 0.05 M (Table 1, entry 8: 61%). Meanwhile, further reducing the catalyst loading from 5 mol % to 2 mol % resulted in a significant decrease in yield (Table 1, entry 9: 28%). When the reaction was scaled up from 0.1 mmol to 0.5 mmol of 1a in consideration of the dead volume of the flow reactor, the product 3a was obtained in markedly improved yield (Table 1, entry 10: 77%) [59] which was comparable to that of the batch reaction (76%). Notably, however, the present flow reaction was performed at 25 °C (batch: 50 °C) with half the amount of AgNTf2 (flow: 5 mol %, batch: 10 mol %), and the light irradiation time was shortened to only 2.5 minutes (batch: 1 h). Thus, under the optimal conditions (Table 1, entry 10), the flow reaction system proved extremely useful for improving the efficiency of the present photochemical sequential transformation.
Table 1: Screening of reaction conditions in the flow reaction systema.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-173-i3.svg?max-width=637&scale=1.0)
|
|||||||
| Entry |
AgNTf2
(x mol %) |
P(4-CF3C6H4)3
(y mol %) |
Conc. of 1a
(z M) |
Flow rate
(R mL/h) |
Premixing
zone (mL) |
Yield of 3a
(%)b |
Recovery of 1a (%)b |
| 1c | 10 | 20 | 0.1 | 3 | none | 42 | 0 |
| 2 | 10 | 20 | 0.1 | 3 | none | 35 | 22 |
| 3 | 10 | 5 | 0.1 | 3 | none | 53 | 1 |
| 4 | 10 | 5 | 0.1 | 24 | none | 53 | 0 |
| 5 | 10 | 5 | 0.1 | 24 | 0.7 | 52 | 9 |
| 6 | 5 | 2.5 | 0.1 | 24 | none | 26 | 14 |
| 7 | 5 | 2.5 | 0.1 | 24 | 0.7 | 49 | 0 |
| 8 | 5 | 2.5 | 0.05 | 24 | 0.7 | 61 | 0 |
| 9 | 2 | 1 | 0.05 | 24 | 0.7 | 28 | 28 |
| 10d | 5 | 2.5 | 0.05 | 24 | 0.7 | 77 | 0 |
aUnless otherwise noted, all reactions were carried out in a flow photochemical reactor (volume: 1.0 mL, λmax = 450 nm) using a dual syringe system, as shown in the table scheme. Syringe A: 0.1 mmol of 1a in 1,2-DCE (0.55 mL). Syringe B: AgNTf2, P(4-CF3C6H4)3, TMSBn (2a), and TFA in 1,2-DCE (0.45 mL); bYield was determined by NMR analysis using 1,1,2,2-tetrabromoethane as an internal standard; cAt 50 °C. dReaction was conducted on a 0.5 mmol scale. Syringe A: 0.5 mmol of 1a in 1,2-DCE (5.4 mL). Syringe B: 25 μmol (5 mol %) of AgNTf2, 12.5 μmol (2.5 mol %) of P(4-CF3C6H4)3, 5 mmol (10 equiv) of TMSBn (2a), and 2.5 mmol (5 equiv) of TFA in 1,2-DCE (4.6 mL).
With the optimal flow reaction conditions in hand, we next investigated the scope of substrates 1 by introducing a series of substituents to the terminal phenyl group. The results of the batch reaction system are also shown for comparison in Table 2 (right-hand side) [55]. As expected, the use of the flow reaction system significantly increased yields, although the yields obtained in the reactions of substrates having an electron-donating methoxy group were low to moderate regardless of the substitution pattern (Table 2, entries 1, 2, and 6). Indeed, when a methoxy group was introduced at the para-position, product 3b was obtained in low yield (Table 2, entry 1: 14%). Because this yield was lower than that obtained in the batch reaction (48%), the flow reaction conditions for 1b were thoroughly reconsidered (see Supporting Information File 1 for details). As a result, extending the premixing time and the light irradiation time (Table 2, entry 2) led to an improved yield; the obtained yield was higher than that of the batch reaction system even when half the amount of AgNTf2 was used with the temperature reduced to 25 °C (flow: 54% vs batch: 48%). Meanwhile, the reaction of substrate 1c having a methyl group as a weak electron-donating group at the para-position afforded product 3c in high yield (Table 2, entry 3: 76%). In addition, the reaction of 1d having a bromo group resulted in a moderate yield, but with a significant improvement compared with the batch reaction (Table 2, entry 4: 57% vs 10%). The reaction of 1e substituted with a strong electron-withdrawing trifluoromethyl group afforded the product 3e in high yield (Table 2, entry 5: 77%), again confirming the high efficiency of the flow reaction system (batch: 54%). Next, the effects of the substituent at the meta-position were investigated. Substrate 1f having a methoxy group afforded compound 3f in only moderate yield (Table 2, entry 6: 39%), similar to the batch reaction (40%). The reactions of substrates having a methyl, bromo, or trifluoromethyl group gave the corresponding products 3g–i, respectively, in good yields (Table 2, entries 7–9). The ortho-methyl-substituted substrate 1j was also compatible, affording product 3j in good yield (Table 2, entry 10: 76%). This yield was comparable to that of the substrate having a methyl group at the para- or meta-position, despite the steric hindrance of the ortho-substituent (Table 2, entry 10 vs entries 3 and 7). When a fluoro group was introduced to the tethering phenyl backbone, a high yield was obtained regardless of whether it was introduced at the 6- or 7-position (Table 2, entries 11 and 12).
Table 2: Scope of substratesa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-173-i4.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | 1 | R1 | R2 | R3 | R4 | R5 | 3 |
Yield of 3
(%)b |
Batch reaction using AgNTf2. Yield of 3 (%)b,c |
| 1 | 1b | MeO | H | H | H | H | 3b | 14 | 48d |
| 2e | 1b | 3b | 54 | ||||||
| 3 | 1c | Me | H | H | H | H | 3c | 76 | 21 |
| 4 | 1d | Br | H | H | H | H | 3d | 57 | 10 |
| 5 | 1e | CF3 | H | H | H | H | 3e | 77 | 65 |
| 6 | 1f | H | MeO | H | H | H | 3f | 39 | 40 |
| 7 | 1g | H | Me | H | H | H | 3g | 75 | 50 |
| 8 | 1h | H | Br | H | H | H | 3h | 68 | 63 |
| 9 | 1i | H | CF3 | H | H | H | 3i | 65 | 42 |
| 10 | 1j | H | H | Me | H | H | 3j | 76 | 72 |
| 11 | 1k | H | H | H | F | H | 3k | 79 | – |
| 12 | 1l | H | H | H | H | F | 3l | 75 | – |
aUnless otherwise noted, all reactions were carried out in the flow photochemical reactor (volume: 1.0 mL, λmax = 450 nm) having a 0.7 mL premixing zone using a dual syringe system with a flow rate of 24 mL/h (12 mL/h for each syringe) at 25 °C. Syringe A: 0.5 mmol of 1 in 1,2-DCE (5.4 mL). Syringe B: 25 μmol (5 mol %) of AgNTf2, 12.5 μmol (2.5 mol %) of P(4-CF3C6H4)3, 5 mmol (10 equiv) of TMSBn (2a), and 2.5 mmol (5 equiv) of TFA in 1,2-DCE (4.6 mL). bYield was determined by NMR analysis using 1,1,2,2-tetrabromoethane as an internal standard. Substrates 1 were not recovered in all cases. All products 3 were isolated before structural assignment. cBatch reaction conditions (see ref. [55]): Unless otherwise noted, all reactions were carried out using blue LED (λmax = 448 nm), 0.1 mmol of 1, 1.0 mmol (10 equiv) of TMSBn (2a), 10 μmol (10 mol %) of AgNTf2, 20 μmol (20 mol %) of P(4-CF3C6H4)3, and 5 equiv of TFA in 1,2-DCE (1.0 mL: 0.1 M of 1) at 50 °C for 1 h. dAt 0 °C for 6 h. eThe flow photochemical reactor having a 1.1 mL premixing zone using a dual syringe system with a flow rate of 6 mL/h (3 mL/h for each syringe).
Next, the effects of a carbonyl substituent, instead of a methyl ketone substituent, were investigated (Table 3). First, the reaction was performed with phenyl ketone 1m, but product 3m was obtained in low yield (Table 3, entry 1: 7%). This yield was lower than that obtained in the batch reaction (30%), and because 28% of 1m were recovered, the flow reaction conditions were further examined (see Supporting Information File 1 for details). Although the yield of 3m was improved to 19% (Table 3, entry 2) by increasing the temperature of the premixing zone from room temperature to 50 °C and reducing the flow rate from 24 mL/h to 6 mL/h (light irradiation time was extended from 2.5 min to 10 min), it did not exceed the yield of the batch reaction. In contrast, aldehyde 1n having a simple phenyl group gave product 3n in good yield (Table 3, entry 3: 72%). Because the yield of this flow reaction was better than that of the batch reaction (65%), the reactions of aldehydes with a series of substituents introduced to the terminal phenyl group were further investigated (Table 3, entries 4–7). Aldehydes having an electron-donating methyl group and an electron-withdrawing bromo group at the para-position of the phenyl moiety gave products 3o and 3p, respectively, in high yields (Table 3, entries 4 and 5). The aldehyde 1q bearing a strong electron-withdrawing trifluoromethyl group at the para-position gave product 3q in moderate yield (Table 3, entry 6: 63%), with the recovery of substrate 1q (18%). The reaction of ortho-methyl-substituted aldehyde 1r afforded the product 3r in high yield when the temperature of the premixing zone was increased to 50 °C (Table 3, entry 7: 73%).
Table 3: Sequential transformation of phenyl ketone 1m and aldehydes 1n–ra.
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-173-i5.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | 1 | R1 | R2 | R3 | 3 | Yield of 3 (%)b | Recovery of 1 (%)b | Batch reaction using AgNTf2. Yield of 3 (%)c |
| 1 | 1m | Ph | H | H | 3m | 7 | 29 | 30 |
| 2d,e | 1m | 3m | 19 | 0 | ||||
| 3 | 1n | H | H | H | 3n | 72 | 0 | 65f |
| 4 | 1o | H | Me | H | 3o | 82 | 5 | – |
| 5 | 1p | H | Br | H | 3p | 75 | 0 | 34f |
| 6 | 1q | H | CF3 | H | 3q | 63 | 18 | – |
| 7e | 1r | H | H | Me | 3r | 73 | 8 | – |
aUnless otherwise noted, all reactions were carried out in the flow photochemical reactor (volume: 1.0 mL, λmax = 450 nm) having a 0.7 mL premixing zone using a dual syringe system with a flow rate of 24 mL/h (12 mL/h for each syringe) at 25 °C. Syringe A: 0.5 mmol of 1 in 1,2-DCE (5.4 mL). Syringe B: 25 μmol (5 mol %) of AgNTf2, 12.5 μmol (2.5 mol %) of P(4-CF3C6H4)3, 5 mmol (10 equiv) of TMSBn (2a), and 2.5 mmol (5 equiv) of TFA in 1,2-DCE (4.6 mL). bYield was determined by NMR analysis using 1,1,2,2-tetrabromoethane as an internal standard. All products 3 were isolated before structural assignment. cBatch reaction conditions: unless otherwise noted, reactions were carried out using 0.1 mmol of 1, 1.0 mmol (10 equiv) of TMSBn (2a), 10 μmol (10 mol %) of AgNTf2, 20 μmol (20 mol %) of P(4-CF3C6H4)3, and 5 equiv of TFA in 1,2-DCE (1 mL: 0.1 M of 1) at 50 °C for 1 h. dThe flow photochemical reactor having a 0.5 mL premixing zone using a dual syringe system with a flow rate of 6 mL/h (3 mL/h for each syringe). eThe temperature of the premixing zone was increased to 50 °C. fThe reaction was performed using 0.05 M of 1 and 2.5 equiv of TFA for 2 h.
The scope of donor molecules 2b and 2c having an electron-withdrawing trifluoromethyl group and an electron-donating methoxy group [60,61] at the para-position of the benzyltrimethylsilane, respectively, was also investigated in the present flow reaction system (Scheme 2). As expected, the flow reaction of 2b having a trifluoromethyl group afforded product 3s in higher yield (50%) than that of the batch reaction (18%) under the optimal reaction conditions. In contrast, in the flow reaction of 2c having a methoxy group, product 3t was obtained in a markedly lower yield (32%) than that of the batch reaction (54%). However, extending the light irradiation time by reducing the flow rate from 24 mL/h to 6 mL/h (light irradiation time: 24 mL/h = 2.5 min, 6 mL/h = 10 min) significantly improved the yield of 3t (78%), presumably because of the retardation of the desilylation process (from B to C in Scheme 1a).
![[1860-5397-20-173-i2]](/bjoc/content/inline/1860-5397-20-173-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The reaction with benzyltrimethylsilane derivatives 2. aFlow rate was 6 mL/h and premixing zone was 0.2 mL.
Scheme 2: The reaction with benzyltrimethylsilane derivatives 2. aFlow rate was 6 mL/h and premixing zone was...
Conclusion
We have demonstrated a flow reaction system for a π-Lewis acidic metal-catalyzed cyclization/photochemical radical addition sequence, affording, in most cases, the 1H-isochromene derivatives in higher yields than the batch reaction system, even with the amount of the π-Lewis acidic metal catalyst reduced by half. In the present sequential transformation, the key cationic species, 2-benzopyrylium intermediates, were generated in situ through the AgNTf2-catalyzed intramolecular cyclization of ortho-carbonyl alkynylbenzene derivatives and subsequent proto-demetalation with TFA. Further photoreactions of 2-benzopyrylium intermediates with benzyltrimethylsilane derivatives as the donor molecule were conducted in the flow photoreactor. We confirmed that the flow reaction system is an excellent method for improving the efficiency of the present sequential transformation, avoiding product degradation under photochemical reaction conditions. Further investigation of other flow photochemical reactions using in situ-generated organic cations is in progress in our laboratory.
Funding
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) and a Grant-in-Aid for Transformative Research Areas (A) “Green Catalysis Science for Renovating Transformation of Carbon-Based Resources” (JP23H04908) from MEXT, Japan and a Grant-in-Aid for Young Scientists (JP19K15552) from JSPS.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors; Wiley-VCH: Weinheim, Germany, 2000. doi:10.1002/3527601953
Return to citation in text: [1] -
Yoshida, J. Basics of Flow Microreactor Synthesis; Springer: Tokyo, Japan, 2015. doi:10.1007/978-4-431-55513-1
Return to citation in text: [1] -
Noël, T., Ed. Organometallic Flow Chemistry; Topics in Organometallic Chemistry, Vol. 57; Springer International Publishing: Cham, Switzerland, 2016. doi:10.1007/978-3-319-33243-7
Return to citation in text: [1] -
Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c
Return to citation in text: [1] -
Ahmed-Omer, B.; Brandt, J. C.; Wirth, T. Org. Biomol. Chem. 2007, 5, 733–740. doi:10.1039/b615072a
Return to citation in text: [1] -
Watts, P.; Wiles, C. Chem. Commun. 2007, 443–467. doi:10.1039/b609428g
Return to citation in text: [1] -
McMullen, J. P.; Jensen, K. F. Annu. Rev. Anal. Chem. 2010, 3, 19–42. doi:10.1146/annurev.anchem.111808.073718
Return to citation in text: [1] -
Yoshida, J.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331–340. doi:10.1002/cssc.201000271
Return to citation in text: [1] -
Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/c1gc16022b
Return to citation in text: [1] -
Kirschning, A.; Kupracz, L.; Hartwig, J. Chem. Lett. 2012, 41, 562–570. doi:10.1246/cl.2012.562
Return to citation in text: [1] -
McQuade, D. T.; Seeberger, P. H. J. Org. Chem. 2013, 78, 6384–6389. doi:10.1021/jo400583m
Return to citation in text: [1] -
Elvira, K. S.; i Solvas, X. C.; Wootton, R. C. R.; deMello, A. J. Nat. Chem. 2013, 5, 905–915. doi:10.1038/nchem.1753
Return to citation in text: [1] -
Pastre, J. C.; Browne, D. L.; Ley, S. V. Chem. Soc. Rev. 2013, 42, 8849–8869. doi:10.1039/c3cs60246j
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183
Return to citation in text: [1] -
Buglioni, L.; Raymenants, F.; Slattery, A.; Zondag, S. D. A.; Noël, T. Chem. Rev. 2022, 122, 2752–2906. doi:10.1021/acs.chemrev.1c00332
Return to citation in text: [1] -
Nagaki, A.; Kim, H.; Yoshida, J. Angew. Chem., Int. Ed. 2008, 47, 7833–7836. doi:10.1002/anie.200803205
Return to citation in text: [1] -
Kim, H.; Min, K.-I.; Inoue, K.; Im, D. J.; Kim, D.-P.; Yoshida, J. Science 2016, 352, 691–694. doi:10.1126/science.aaf1389
Return to citation in text: [1] -
Seo, H.; Katcher, M. H.; Jamison, T. F. Nat. Chem. 2017, 9, 453–456. doi:10.1038/nchem.2690
Return to citation in text: [1] -
Otake, Y.; Nakamura, H.; Fuse, S. Angew. Chem., Int. Ed. 2018, 57, 11389–11393. doi:10.1002/anie.201803549
Return to citation in text: [1] -
Nagaki, A.; Yamashita, H.; Hirose, K.; Tsuchihashi, Y.; Yoshida, J. Angew. Chem., Int. Ed. 2019, 58, 4027–4030. doi:10.1002/anie.201814088
Return to citation in text: [1] -
Nagaki, A.; Ichinari, D.; Yoshida, J. Chem. Commun. 2013, 49, 3242–3244. doi:10.1039/c3cc40392k
Return to citation in text: [1] -
Moon, S.-Y.; Jung, S.-H.; Bin Kim, U.; Kim, W.-S. RSC Adv. 2015, 5, 79385–79390. doi:10.1039/c5ra14890a
Return to citation in text: [1] -
Degennaro, L.; Maggiulli, D.; Carlucci, C.; Fanelli, F.; Romanazzi, G.; Luisi, R. Chem. Commun. 2016, 52, 9554–9557. doi:10.1039/c6cc04588j
Return to citation in text: [1] -
Nauth, A. M.; Lipp, A.; Lipp, B.; Opatz, T. Eur. J. Org. Chem. 2017, 2099–2103. doi:10.1002/ejoc.201601394
Return to citation in text: [1] -
Tucker, J. W.; Zhang, Y.; Jamison, T. F.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. doi:10.1002/anie.201200961
Return to citation in text: [1] -
Hernandez‐Perez, A. C.; Collins, S. K. Angew. Chem., Int. Ed. 2013, 52, 12696–12700. doi:10.1002/anie.201306920
Return to citation in text: [1] -
Elliott, L. D.; Knowles, J. P.; Koovits, P. J.; Maskill, K. G.; Ralph, M. J.; Lejeune, G.; Edwards, L. J.; Robinson, R. I.; Clemens, I. R.; Cox, B.; Pascoe, D. D.; Koch, G.; Eberle, M.; Berry, M. B.; Booker‐Milburn, K. I. Chem. – Eur. J. 2014, 20, 15226–15232. doi:10.1002/chem.201404347
Return to citation in text: [1] -
Talla, A.; Driessen, B.; Straathof, N. J. W.; Milroy, L.-G.; Brunsveld, L.; Hessel, V.; Noël, T. Adv. Synth. Catal. 2015, 357, 2180–2186. doi:10.1002/adsc.201401010
Return to citation in text: [1] -
Asao, N.; Nogami, T.; Takahashi, K.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 764–765. doi:10.1021/ja017366b
Return to citation in text: [1] -
Yao, T.; Zhang, X.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 11164–11165. doi:10.1021/ja0466964
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536–6544. doi:10.1039/c1cc10780a
Return to citation in text: [1] -
Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d
Return to citation in text: [1] -
Saito, K.; Kajiwara, Y.; Akiyama, T. Angew. Chem., Int. Ed. 2013, 52, 13284–13288. doi:10.1002/anie.201308303
Return to citation in text: [1] -
Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a
Return to citation in text: [1] -
Terada, M.; Li, F.; Toda, Y. Angew. Chem., Int. Ed. 2014, 53, 235–239. doi:10.1002/anie.201307371
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Debrouwer, W.; Heugebaert, T. S. A.; Roman, B. I.; Stevens, C. V. Adv. Synth. Catal. 2015, 357, 2975–3006. doi:10.1002/adsc.201500520
Return to citation in text: [1] -
Asiri, A. M.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 4471–4503. doi:10.1039/c6cs00023a
Return to citation in text: [1] -
Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Chem. Rev. 2017, 117, 14091–14200. doi:10.1021/acs.chemrev.7b00343
Return to citation in text: [1] -
Zhang, Z.; Smal, V.; Retailleau, P.; Voituriez, A.; Frison, G.; Marinetti, A.; Guinchard, X. J. Am. Chem. Soc. 2020, 142, 3797–3805. doi:10.1021/jacs.9b11154
Return to citation in text: [1] -
Raj, A. S. K.; Narode, A. S.; Liu, R.-S. Org. Lett. 2021, 23, 1378–1382. doi:10.1021/acs.orglett.1c00038
Return to citation in text: [1] -
Greiner, L. C.; Arichi, N.; Inuki, S.; Ohno, H. Angew. Chem., Int. Ed. 2023, 62, e202213653. doi:10.1002/anie.202213653
Return to citation in text: [1] -
Das, S. Asian J. Org. Chem. 2023, 12, e202300267. doi:10.1002/ajoc.202300267
Return to citation in text: [1] -
Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi:10.1038/ncomms8919
Return to citation in text: [1] -
Nakajima, K.; Miyake, Y.; Nishibayashi, Y. Acc. Chem. Res. 2016, 49, 1946–1956. doi:10.1021/acs.accounts.6b00251
Return to citation in text: [1] -
Ermanis, K.; Colgan, A. C.; Proctor, R. S. J.; Hadrys, B. W.; Phipps, R. J.; Goodman, J. M. J. Am. Chem. Soc. 2020, 142, 21091–21101. doi:10.1021/jacs.0c09668
Return to citation in text: [1] -
Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2021, 50, 8857–8873. doi:10.1039/d1cs00208b
Return to citation in text: [1] -
Alfonzo, E.; Hande, S. M. ACS Catal. 2020, 10, 12590–12595. doi:10.1021/acscatal.0c03851
Return to citation in text: [1] -
Kikuchi, J.; Kodama, S.; Terada, M. Org. Chem. Front. 2021, 8, 4153–4159. doi:10.1039/d1qo00657f
Return to citation in text: [1] -
Schlegel, M.; Qian, S.; Nicewicz, D. A. ACS Catal. 2022, 12, 10499–10505. doi:10.1021/acscatal.2c02997
Return to citation in text: [1] -
Takemura, N.; Sumida, Y.; Ohmiya, H. ACS Catal. 2022, 12, 7804–7810. doi:10.1021/acscatal.2c01964
Return to citation in text: [1] -
Miura, T.; Yoritate, M.; Hirai, G. Chem. Commun. 2023, 59, 8564–8567. doi:10.1039/d3cc02361c
Return to citation in text: [1] -
Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a
Return to citation in text: [1] [2] [3] [4] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057
Return to citation in text: [1] -
Cu(NTf2)2 which was also the effective catalyst in our batch system (ref. [55]) did not solve well in 1,2-DCE.
Return to citation in text: [1] -
Benzyltrimethylsilane (2a), which was not consumed in the corresponding radical reaction, was almost completely recovered.
Return to citation in text: [1] -
Maruyama, T.; Mizuno, Y.; Shimizu, I.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2007, 129, 1902–1903. doi:10.1021/ja068589a
Return to citation in text: [1] -
Montanaro, S.; Ravelli, D.; Merli, D.; Fagnoni, M.; Albini, A. Org. Lett. 2012, 14, 4218–4221. doi:10.1021/ol301900p
Return to citation in text: [1]
| 1. | Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors; Wiley-VCH: Weinheim, Germany, 2000. doi:10.1002/3527601953 |
| 2. | Yoshida, J. Basics of Flow Microreactor Synthesis; Springer: Tokyo, Japan, 2015. doi:10.1007/978-4-431-55513-1 |
| 3. | Noël, T., Ed. Organometallic Flow Chemistry; Topics in Organometallic Chemistry, Vol. 57; Springer International Publishing: Cham, Switzerland, 2016. doi:10.1007/978-3-319-33243-7 |
| 4. | Mason, B. P.; Price, K. E.; Steinbacher, J. L.; Bogdan, A. R.; McQuade, D. T. Chem. Rev. 2007, 107, 2300–2318. doi:10.1021/cr050944c |
| 5. | Ahmed-Omer, B.; Brandt, J. C.; Wirth, T. Org. Biomol. Chem. 2007, 5, 733–740. doi:10.1039/b615072a |
| 6. | Watts, P.; Wiles, C. Chem. Commun. 2007, 443–467. doi:10.1039/b609428g |
| 7. | McMullen, J. P.; Jensen, K. F. Annu. Rev. Anal. Chem. 2010, 3, 19–42. doi:10.1146/annurev.anchem.111808.073718 |
| 8. | Yoshida, J.; Kim, H.; Nagaki, A. ChemSusChem 2011, 4, 331–340. doi:10.1002/cssc.201000271 |
| 9. | Wiles, C.; Watts, P. Green Chem. 2012, 14, 38–54. doi:10.1039/c1gc16022b |
| 10. | Kirschning, A.; Kupracz, L.; Hartwig, J. Chem. Lett. 2012, 41, 562–570. doi:10.1246/cl.2012.562 |
| 11. | McQuade, D. T.; Seeberger, P. H. J. Org. Chem. 2013, 78, 6384–6389. doi:10.1021/jo400583m |
| 12. | Elvira, K. S.; i Solvas, X. C.; Wootton, R. C. R.; deMello, A. J. Nat. Chem. 2013, 5, 905–915. doi:10.1038/nchem.1753 |
| 13. | Pastre, J. C.; Browne, D. L.; Ley, S. V. Chem. Soc. Rev. 2013, 42, 8849–8869. doi:10.1039/c3cs60246j |
| 14. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 15. | Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183 |
| 16. | Buglioni, L.; Raymenants, F.; Slattery, A.; Zondag, S. D. A.; Noël, T. Chem. Rev. 2022, 122, 2752–2906. doi:10.1021/acs.chemrev.1c00332 |
| 30. | Asao, N.; Nogami, T.; Takahashi, K.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 764–765. doi:10.1021/ja017366b |
| 31. | Yao, T.; Zhang, X.; Larock, R. C. J. Am. Chem. Soc. 2004, 126, 11164–11165. doi:10.1021/ja0466964 |
| 32. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 33. | Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536–6544. doi:10.1039/c1cc10780a |
| 34. | Shiroodi, R. K.; Gevorgyan, V. Chem. Soc. Rev. 2013, 42, 4991–5001. doi:10.1039/c3cs35514d |
| 35. | Saito, K.; Kajiwara, Y.; Akiyama, T. Angew. Chem., Int. Ed. 2013, 52, 13284–13288. doi:10.1002/anie.201308303 |
| 36. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a |
| 37. | Terada, M.; Li, F.; Toda, Y. Angew. Chem., Int. Ed. 2014, 53, 235–239. doi:10.1002/anie.201307371 |
| 38. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 39. | Debrouwer, W.; Heugebaert, T. S. A.; Roman, B. I.; Stevens, C. V. Adv. Synth. Catal. 2015, 357, 2975–3006. doi:10.1002/adsc.201500520 |
| 40. | Asiri, A. M.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 4471–4503. doi:10.1039/c6cs00023a |
| 41. | Lauder, K.; Toscani, A.; Scalacci, N.; Castagnolo, D. Chem. Rev. 2017, 117, 14091–14200. doi:10.1021/acs.chemrev.7b00343 |
| 42. | Zhang, Z.; Smal, V.; Retailleau, P.; Voituriez, A.; Frison, G.; Marinetti, A.; Guinchard, X. J. Am. Chem. Soc. 2020, 142, 3797–3805. doi:10.1021/jacs.9b11154 |
| 43. | Raj, A. S. K.; Narode, A. S.; Liu, R.-S. Org. Lett. 2021, 23, 1378–1382. doi:10.1021/acs.orglett.1c00038 |
| 44. | Greiner, L. C.; Arichi, N.; Inuki, S.; Ohno, H. Angew. Chem., Int. Ed. 2023, 62, e202213653. doi:10.1002/anie.202213653 |
| 45. | Das, S. Asian J. Org. Chem. 2023, 12, e202300267. doi:10.1002/ajoc.202300267 |
| 55. | Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a |
| 26. | Tucker, J. W.; Zhang, Y.; Jamison, T. F.; Stephenson, C. R. J. Angew. Chem., Int. Ed. 2012, 51, 4144–4147. doi:10.1002/anie.201200961 |
| 27. | Hernandez‐Perez, A. C.; Collins, S. K. Angew. Chem., Int. Ed. 2013, 52, 12696–12700. doi:10.1002/anie.201306920 |
| 28. | Elliott, L. D.; Knowles, J. P.; Koovits, P. J.; Maskill, K. G.; Ralph, M. J.; Lejeune, G.; Edwards, L. J.; Robinson, R. I.; Clemens, I. R.; Cox, B.; Pascoe, D. D.; Koch, G.; Eberle, M.; Berry, M. B.; Booker‐Milburn, K. I. Chem. – Eur. J. 2014, 20, 15226–15232. doi:10.1002/chem.201404347 |
| 29. | Talla, A.; Driessen, B.; Straathof, N. J. W.; Milroy, L.-G.; Brunsveld, L.; Hessel, V.; Noël, T. Adv. Synth. Catal. 2015, 357, 2180–2186. doi:10.1002/adsc.201401010 |
| 22. | Nagaki, A.; Ichinari, D.; Yoshida, J. Chem. Commun. 2013, 49, 3242–3244. doi:10.1039/c3cc40392k |
| 23. | Moon, S.-Y.; Jung, S.-H.; Bin Kim, U.; Kim, W.-S. RSC Adv. 2015, 5, 79385–79390. doi:10.1039/c5ra14890a |
| 24. | Degennaro, L.; Maggiulli, D.; Carlucci, C.; Fanelli, F.; Romanazzi, G.; Luisi, R. Chem. Commun. 2016, 52, 9554–9557. doi:10.1039/c6cc04588j |
| 25. | Nauth, A. M.; Lipp, A.; Lipp, B.; Opatz, T. Eur. J. Org. Chem. 2017, 2099–2103. doi:10.1002/ejoc.201601394 |
| 55. | Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a |
| 17. | Nagaki, A.; Kim, H.; Yoshida, J. Angew. Chem., Int. Ed. 2008, 47, 7833–7836. doi:10.1002/anie.200803205 |
| 18. | Kim, H.; Min, K.-I.; Inoue, K.; Im, D. J.; Kim, D.-P.; Yoshida, J. Science 2016, 352, 691–694. doi:10.1126/science.aaf1389 |
| 19. | Seo, H.; Katcher, M. H.; Jamison, T. F. Nat. Chem. 2017, 9, 453–456. doi:10.1038/nchem.2690 |
| 20. | Otake, Y.; Nakamura, H.; Fuse, S. Angew. Chem., Int. Ed. 2018, 57, 11389–11393. doi:10.1002/anie.201803549 |
| 21. | Nagaki, A.; Yamashita, H.; Hirose, K.; Tsuchihashi, Y.; Yoshida, J. Angew. Chem., Int. Ed. 2019, 58, 4027–4030. doi:10.1002/anie.201814088 |
| 60. | Maruyama, T.; Mizuno, Y.; Shimizu, I.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2007, 129, 1902–1903. doi:10.1021/ja068589a |
| 61. | Montanaro, S.; Ravelli, D.; Merli, D.; Fagnoni, M.; Albini, A. Org. Lett. 2012, 14, 4218–4221. doi:10.1021/ol301900p |
| 58. | Cu(NTf2)2 which was also the effective catalyst in our batch system (ref. [55]) did not solve well in 1,2-DCE. |
| 59. | Benzyltrimethylsilane (2a), which was not consumed in the corresponding radical reaction, was almost completely recovered. |
| 56. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 57. | Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057 |
| 55. | Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a |
| 55. | Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a |
| 46. | Beatty, J. W.; Douglas, J. J.; Cole, K. P.; Stephenson, C. R. J. Nat. Commun. 2015, 6, 7919. doi:10.1038/ncomms8919 |
| 47. | Nakajima, K.; Miyake, Y.; Nishibayashi, Y. Acc. Chem. Res. 2016, 49, 1946–1956. doi:10.1021/acs.accounts.6b00251 |
| 48. | Ermanis, K.; Colgan, A. C.; Proctor, R. S. J.; Hadrys, B. W.; Phipps, R. J.; Goodman, J. M. J. Am. Chem. Soc. 2020, 142, 21091–21101. doi:10.1021/jacs.0c09668 |
| 49. | Xiong, T.; Zhang, Q. Chem. Soc. Rev. 2021, 50, 8857–8873. doi:10.1039/d1cs00208b |
| 50. | Alfonzo, E.; Hande, S. M. ACS Catal. 2020, 10, 12590–12595. doi:10.1021/acscatal.0c03851 |
| 51. | Kikuchi, J.; Kodama, S.; Terada, M. Org. Chem. Front. 2021, 8, 4153–4159. doi:10.1039/d1qo00657f |
| 52. | Schlegel, M.; Qian, S.; Nicewicz, D. A. ACS Catal. 2022, 12, 10499–10505. doi:10.1021/acscatal.2c02997 |
| 53. | Takemura, N.; Sumida, Y.; Ohmiya, H. ACS Catal. 2022, 12, 7804–7810. doi:10.1021/acscatal.2c01964 |
| 54. | Miura, T.; Yoritate, M.; Hirai, G. Chem. Commun. 2023, 59, 8564–8567. doi:10.1039/d3cc02361c |
| 55. | Terada, M.; Yazaki, R.; Obayashi, R.; Iwasaki, Z.; Umemiya, S.; Kikuchi, J. Chem. Sci. 2024, 15, 6115–6121. doi:10.1039/d4sc00808a |
© 2024 Terada et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.