1Pharmacognosy Department, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
2Biotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, 5180 Kurokawa, Imizu, Toyama 939-0398, Japan
3Computational Chemistry Lab, Department of Chemistry, Faculty of Science, Ain Shams University, 11566 Abbassia, Cairo, Egypt
Corresponding author email
Associate Editor: S. Bräse Beilstein J. Org. Chem.2024,20, 1981–1987.https://doi.org/10.3762/bjoc.20.174 Received 24 Apr 2024,
Accepted 29 Jul 2024,
Published 13 Aug 2024
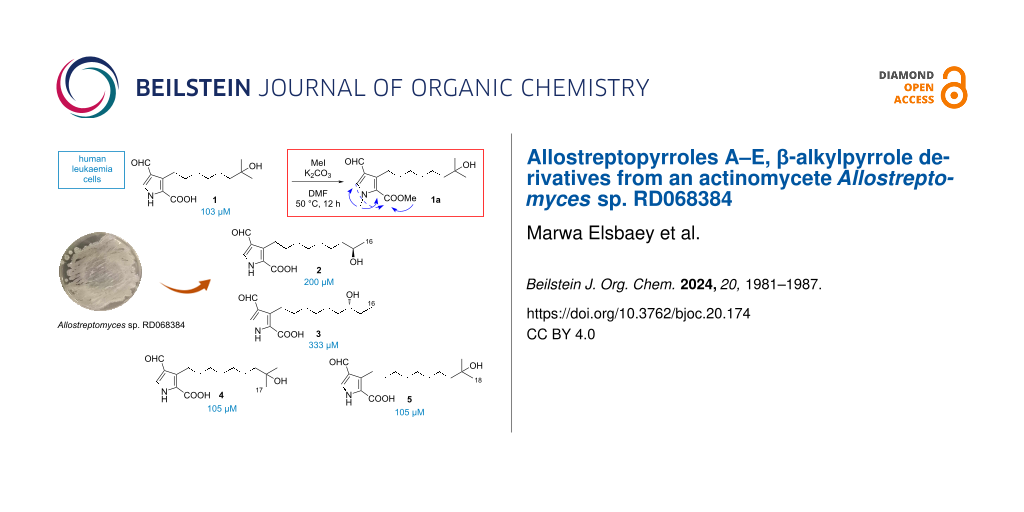
Five new β-alkylpyrrole derivatives, allostreptopyrroles A–E (1–5), were isolated from the culture broth of Allostreptomyces RD068384. Their structures were elucidated by 1D and 2D NMR spectroscopic analyses, HRESIMS, and chemical derivatization. The absolute configurations of compounds 2 and 3 were predicted by comparison of experimental and calculated specific rotation data. Compounds 1–5 are the first examples of natural pyrroles substituted by formyl and carboxyl functionalities. Compounds 1, 4, and 5 showed cytotoxicity against Kasumi-1 human acute myeloblastic leukemia cells with IC50 values of 103, 105, and 105 μM, respectively, which are less active than the anticancer agent cisplatin, with an IC50 value of 70 μM.
β-Alkylpyrroles are key structural motifs in biomolecules and functional organic materials [1]. For instance, β-alkylpyrroles are the main building blocks for the life-essential tetrapyrrole pigments (porphyrins) including heme, chlorophyll, and vitamin B12 [1,2] (Figure S54 in Supporting Information File 1). Porphobilinogen, the fundamental biological precursor of tetrapyrroles, is biosynthesized via asymmetric condensation of two δ-aminolevulinic acid molecules [2,3]. From another aspect, copolymerized β-alkylpyrroles are among the most investigated organic materials for their enhanced physical and electrochemical properties [4,5]. Accordingly, chemists have focused on developing selective synthetic strategies for the construction of β-alkylpyrroles [1].
While the pyrrole nucleus is featured in many marine natural products [6,7], pyrroles substituted with long hydrocarbon chains (pyrrole lipids) are seldomly isolated, and their presence is limited to certain marine organisms [8]. A series of 3-alkylpyrrole-2-carbaldehydes/carboxylic acid/methylcarboxylate was reported from the marine sponge Oscarella lobularis (Figure 1 and Figure S54 in Supporting Information File 1) [7,9], but the actual position of the alkyl chains is very likely to be on the 5 position, as Stierle and Faulkner pointed in their study on a series of 5-alkylpyrrole-2-carbaldehydes from the sponge Laxosuberites sp. [10]. From 1997 to 2017, over fifty 5-alkylpyrrole-2-carbaldehydes and 5-alkyl-2-hydroxymethylpyrroles with diversely functionalized alkyl side chains have been isolated from sponges of the genus Mycale[7,11], but no additional 3-alkylpyrroles were reported so far.
Figure 1:
Structures of allostreptopyrroles A–E (1–5) and related metabolites.
Figure 1:
Structures of allostreptopyrroles A–E (1–5) and related metabolites.
β-Alkylpyrroles are rare as microbial metabolites, and most of them are pyrroloterpenes from Streptomyces (Figure 1 and Figure S54 in Supporting Information File 1). Examples include pyrrolostatin [12] and its congener geranylpyrrol A [13], bearing a carboxylic group at the C2 and a geranyl group at the C4 position of the pyrrole ring, and their 2-nitro congeners, nitropyrrolins [14] and heronapyrroles [15], bearing a farnesyl chain at the C4 position. Pyrroloterpenes are proposed to be of mixed biogenesis, elaborated from an aromatic pyrrole moiety and a terpenoid chain [15]. Prodigiosin, a major metabolite of Serratia, is another example of β-alkylpyrrole, bearing a pentyl chain on the pyrrolyldipyrromethene core [16]. Similarly, α-alkylpyrroles are limited to a handful examples including α-pyrrolosesquiterpenes [17-19], undecylprodigiosin [16] from Streptomyces, and fungus-derived pyrrol-2-ylpolyenes [20].
In 2017, Allostreptomyces was introduced as a new genus in the family Streptomycetaceae[21], and two species, A. psammosilenae[21] and A. indica[22], are currently known. Only two 22-membered macrolides were reported from this genus [23] until we recently isolated five polycyclic tetramate-class macrolactams from Allostreptomyces sp. RD068384, including a new congener, allostreptamide [24]. Further investigation of this strain led to the isolation of five new β-alkylpyrroles, designated allostreptopyrroles A–E (1–5) (Figure 1).
Results and Discussion
The fermentation extract of strain RD068384, cultured in A-3M medium, was fractionated on a silica gel column eluting with CHCl3/MeOH mixtures. Allostreptamide was obtained from the eluate with CHCl3/MeOH 2:1 [24]. HPLC-DAD analysis of a less polar fraction, eluted with CHCl3/MeOH 10:1, detected several peaks with characteristic UV absorptions, which were purified by ODS flash chromatography followed by ODS HPLC to yield compounds 1–5.
Allostreptopyrrole A (1) was obtained as a greenish yellow amorphous solid. The molecular formula was determined to be C15H23NO4 based on a molecular ion peak at m/z 280.1550 [M − H]− (calcd for 280.1554) observed in a negative HRESITOF mass spectrum. Analysis of 1H NMR, 13C NMR (Table 1), and HSQC spectra revealed a formyl group (δC 186.3/δH 9.89), an olefinic methine (δC 131.1/δH 7.64), an acyl carbonyl carbon (δC 163.3), three non-protonated olefinic carbons (δC 133.4, 126.4, and 123.1), a deshielded non-protonated sp3 carbon (δC 70.1), six sp3 methylenes (δC 25.0–44.8), and two magnetically equivalent tertiary methyl groups (δC 29.5/δH 1.13). These molecular parts accounted for four degrees of unsaturation out of five, leaving one degree for a ring structure. In addition, a highly conjugated functional group was suggested by UV maximal absorptions at 235 nm and 273 nm and HMBC correlations from the formyl and the olefinic methine protons to all sp2 carbons except the acyl carbonyl carbon (Figure 2 and Table S1 in Supporting Information File 1). The sp3 carbons, in contrast, constituted an alkyl chain: the six methylene units were connected in sequence to form a hexamethylene chain as supported by overlapping six proton resonances at δH 1.31–1.42 and by inter-unit COSY and HMBC correlations. This methylene chain was blocked by an oxypropyl group, as evident from HMBC correlations from the tertiary methyl protons (δH 1.13) to the oxygenated carbon (δC 70.1), and one of the methylene carbons (C13: δC 44.8).
Table 1:1H and 13C NMR data for 1 and 1a.a
1
1b
1a
Position
δC
δH, mult, J in Hz
δC
δH, mult, J in Hz
δC
δH, mult, J in Hz
2
123.1
–
122.4
–
122.8
–
3
133.4
–
133.2
–
135.9
–
4
126.4
–
126.4
–
124.0
–
5
131.1
7.64, s
131.6
7.51, s
137.0
7.63, s
6
163.3
–
166.1c
–
162.5
–
7
186.3
9.89, s
188.4
9.77, s
185.6
9.83, s
8
25.4
3.13, t (7.5)
25.7
3.11, t (7.5)
26.0
3.06, t (7.8)
9
32.0
1.59, m
32.3
1.57, m
32.2
1.55, m
10
30.5d
1.39, m
30.6
1.38, me
30.4f
1.28–1.44, m
11
30.9d
1.31, m
31.3
1.28–1.45, m
30.9
1.28–1.44, m
12
25.0
1.33–1.42, m
25.4
1.31–1.44, m
25.0
1.35–1.42, m
13
44.8
1.42, m
44.9
1.44, m
44.9
1.42, m
14
70.1
–
71.5
–
70.1
–
15
29.5f
1.13, s
29.1
1.15, s
29.6f
1.13, s
16
29.5f
1.13, s
29.1
1.15, s
29.6f
1.13, s
NCH3
–
–
161.4g
–
38.5
3.94, s
COOCH3
–
–
–
–
51.4
3.85, s
aNMR data were recorded in CD3COCD3 at 500 and 125 MHz for 1H and 13C, respectively. bRecorded in CD3OD. cAssigned from HMBC. dInterchangeable. eAssigned from COSY. fOverlapping signals read from HSQC. g15N chemical shift determined from 15N HMBC.
Figure 2:
COSY, 15N-HMBC and key HMBC correlations of compounds 1–5 and 1a.
Figure 2:
COSY, 15N-HMBC and key HMBC correlations of compounds 1–5 and 1a.
The formyl proton H7 showed HMBC correlations to the olefinic carbons C3, C4, and C5 and the olefinic methine proton H5 was correlated with C2, C3, C4, and C7. These correlation data allowed the assignment of a carbon sequence C2–C3–C4–C5 and the attachment of the formyl group at C4. Furthermore, HMBC correlations from two methylene protons H28 to the olefinic carbons C2, C3, and C4 connected the chain part at C3. A 1H,15N-HMBC correlation was seen from H5 to a nitrogen at δN 161.4, which suggested the presence of a nitrogen atom adjacent to C5. A correlation to the acyl carbonyl carbon (C6) was not available at this stage. In order to obtain further information for connectivity, compound 1 was reacted with methyl iodide and K2CO3 to give a bismethylated derivative 1a. A methyl proton at δH 3.94 was of an N-methyl group (δC 38.5) and displayed two strong HMBC correlations to C2 and C5, which connected these carbons through a nitrogen atom to establish a pyrrole ring, and also a hydroxy group at the alkyl terminus. Another methyl proton at δH 3.85 was of a methoxy group (δC 51.4) and had only one HMBC correlation to C6, which provided a methoxycarbonyl (–COOMe) fragment. Finally, this fragment was placed at C2 by an HMBC correlation from H5 to C6 to complete the gross structure of 1.
Both compounds 2 and 3 were obtained as greenish yellow amorphous and their molecular formula were suggested to be the same as that of 1 from HRESITOFMS and NMR analytical data (Table 2), inferring that compounds 2 and 3 were isomers of 1. In fact, their NMR spectra were closely similar to those for 1 except a little difference in the alkyl side chain terminus. In a COSY spectrum of 2, the terminal doublet methyl proton was correlated with an oxymethine H15, which in turn was correlated with a methylene H214. The pyrrole moiety with the same substituents as 1 was deduced from HMBC correlations. Therefore, compound 2 was determined to have a non-branched alkyl chain with a hydroxy group at C15. Meanwhile, 3 possessed a terminal ethyl group, which was connected to an oxymethine H14 in a COSY spectrum, thereby establishing a non-branched alkyl chain with a hydroxy group at C14.
Table 2:1H and 13C NMR data for compounds 2 and 3 in CD3COCD3.
2
3
Position
δC
δH, mult, J in Hz
δC
δH, mult, J in Hz
2
123.0
–
122.8
–
3
133.5
–
133.7
–
4
126.5
–
126.5
–
5
131.2
7.64, s
131.3
7.66, s
6
163.0
–
162.9
–
7
186.3
9.89, s
186.3
9.90, s
8
25.3
3.13, t (7.0)
25.4
3.14, brs
9
32.0
1.59, m
32.0
1.59, m
10
30.6a
1.38, mb
30.6c
1.39, mb
11
30.4a
1.25–1.46, m
31.1a
1.26–1.48, m
12
30.5a
1.25–1.46, m
26.5
1.32–1.44, m
13
26.6
1.31–1.41, m
38.0
1.37, m
14
40.3
1.36–1.40, m
72.7
3.42, br
15
67.5
3.68, m
31.0a
1.36, 1.46, mb
16
24.0
1.10, d (6.1)
10.5
0.90, t (7.1)
aInterchangeable. bAssigned from COSY. cOverlapping signals read from HSQC.
The specific rotation values of 2 and 3 were calculated to predict their absolute configurations. For the flexible molecules 2 and 3, thousands of conformers may exist (over 52400 conformers). However, only a few are usually significantly populated (i.e., the compound exists as a rapidly equilibrating mixture of multiple conformers). In this situation, the spectroscopic properties of a molecule can be calculated as the average over the conformers, weighted according to their populations [25]. The calculated specific rotations −11.4 and +16.1 were obtained for R-configured 2 and 3 from the DFT computations (see DFT methodology section), respectively, which were in good agreement with the experimentally obtained values, −6.1 for 2 and +15 for 3. Thus, R-configurations were proposed for compounds 2 and 3. However, this prediction was not confirmed by chemical derivatization due to their limited availability.
1H and 13C NMR spectra of compounds 4 and 5 were superimposable to those of 1 except for methylene resonances, supporting that both 4 and 5 possess the same substituted pyrrole ring and hydroxyisopropyl terminus as compound 1 (Table 3). HRESITOFMS analysis determined the molecular formula of 4 to be C16H25NO4 and that of 5 to be C17H27NO4, which established that 4 and 5 are one- and two-methylene-longer congeners of 1.
Table 3:1H and 13C NMR data for 4 and 5.
4 in CD3COCD3
5 in CD3OD
Position
δC
δH, mult, J in Hz
δC
δH, mult, J in Hz
2
122.4
–
125.2
–
3
133.8
–
133.6
–
4
126.5
–
126.4
–
5
131.4
7.68, s
131.8
7.54, s
6
162.5
–
165.8
–
7
186.3
9.90, s
188.4
9.78, s
8
25.3
3.13, t (7.8)
25.7
3.10, t (7.3)
9
32.0
1.60, m
32.4
1.56, m
10
30.2a
1.38, mb
30.6c
1.39, mb
11
31.1c
1.26–1.44, m
30.7c
1.27–1.46, m
12
31.0c
1.26–1.44, m
30.6c
1.27–1.46, m
13
25.0
1.32–1.41, m
31.4
1.27–1.46, m
14
44.8
1.42, m
25.4
1.31–1.41, m
15
70.1
–
44.9
1.44, m
16
29.5a
1.14, s
71.5
–
17
29.5a
1.14, s
29.1
1.16, s
18
–
–
29.1
1.16, s
N
–
–
162.3d
–
aOverlapping signals read from HSQC. bAssigned from COSY. cInterchangeable. d15N chemical shift determined from 15N HMBC.
Compounds 1, 4, and 5 showed moderate cytotoxicity against Kasumi-1 human acute myeloblastic leukemia cells with IC50 values of 103, 105, and 105 while 2 and 3 were less active with IC50 values of 200 and 333 μM, respectively. Under the same experimental conditions, cisplatin, a positive control, inhibited the cell growth with an IC50 value of 70 μM. Compounds 1–5 were merely inhibitory against tyrosinase, showing 19, 13, 9.6, 18, and 15% inhibition at 200 μM, respectively, while a positive control, kojic acid, inhibited the same enzyme by 95%.
Conclusion
In summary, five new alkylpyrroles, allostreptopyrroles A–E (1–5), were discovered from a fermentation extract of Allostreptomyces sp. RD068384, a strain belonging to an almost unstudied actinomycetes genus within the family Streptomycetaceae.
Compounds 1–5 are characterized by a pyrrole-2-carboxylic acid core decorated with a formyl group and an alkyl side chain. Secondary metabolites of this specific composition have not been reported. The pyrrole-2-carboxyl skeleton is a recurring framework in pyrrolic natural products including microbial pyrrolostatin and aminocoumarin antibiotics [2], plant-derived brachystemidines [26], and lamellarins from marine invertebrates [6] (Figure S55 in Supporting Information File 1). Biosynthetically, pyrrole-2-carboxylic acid is known to be derived from ʟ-proline [2]. Similarly, pyrrole-2-carbaldehydes have been isolated from various natural sources including plants, marine invertebrates, and fungi [7], while 1–5 are the first to have formyl and carboxyl functionalities. Furthermore, a β-alkyl substitution is not very common in pyrrolic secondary metabolites. The most related metabolites to 1–5 are the reported alkylpyrroles from a marine sponge Oscarella lobularis[7] and pyrroloterpenes from Streptomyces[12-15], although the substitution patterns are different (Figure 1). Natural alkylpyrroles were shown to have cytotoxicity [27], antidiabetic activity [28], anti-lipid peroxidation [12], in vivo antihypoxic activity [12], and antibacterial activity [15]. Though not impressive in cytotoxicity and tyrosinase-inhibitory evaluations, compounds 1–5 could be more potent in some other bioassays, which is a subject of future studies. Finally, these results supported that actinomycetes genera with little or no chemical study are a fruitful reservoir for discovering new natural molecules.
Experimental
Microorganism, fermentation, extraction, and isolation
Details on the supplier of Allostreptomyces sp. RD068384, fermentation, extraction, and fractionation are described in Supporting Information File 1. While a CHCl3/MeOH 2:1-eluting fraction by silica gel open column chromatography eventually yielded allostreptamide [24], a less polar CHCl3/MeOH 10:1 fraction contained compounds with characteristic UV absorption. This fraction, obtained as 395 mg of brown solid from 4 L culture in A-3M medium, was fractionated by octadecyldimethylsilyl (ODS) silica gel column chromatography with a gradient of MeCN/0.1% HCO2H solution (2:8, 3:7, 4:6, 5:5, 6:4, 7:3, and 8:2, v/v). The third (4:6) and fourth fractions (5:5) contained the peaks of our target, which were combined (35 mg) and purified by ODS-HPLC (Cosmosil C18 AR-II, 10 × 250 mm, 4 mL/min, UV detection at 254 nm) eluted with 33% MeCN/0.1% HCO2H solution to yield 1 (1.2 mg, tR 10.1 min), 2 (0.9 mg, tR 12.1 min), 3 (0.8 mg, tR 13.7 min), and 4 (2.3 mg, tR 17.3 min). At time 22 min, the MeCN concentration was raised to 35%, which eluted 5 (1.7 mg) at tR 28.9 min. To obtain higher amounts of compounds, fermentation with 4 L culture and isolation were repeated twice to afford in total 6.5 mg of 1, 3.1 mg of 2, 2.6 mg of 3, 7.2 mg of 4, and 5.6 mg of 5 from 12 L culture.
Allostreptopyrrole A (1): greenish yellow amorphous solid; UV (MeOH) λmax nm (log ε) 234 (3.86), 273 sh (3.44); IR (ATR) νmax: 3275, 2964, 2928, 2855, 1658, 1554, 1418 cm−1; 1H and 13C NMR data, see Table 1; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1550.
Allostreptopyrrole B (2): greenish yellow amorphous solid; +15 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.87), 273 sh (3.49); IR (ATR) νmax: 3263, 2964, 2925, 2854, 1658, 1556, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1554.
Allostreptopyrrole C (3): greenish yellow amorphous solid; −6.1 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.82), 276 sh (3.46); IR (ATR) νmax: 3265, 2925, 2856, 1657, 1555, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1556.
Allostreptopyrrole D (4): greenish yellow amorphous solid; UV (MeOH) λmax, nm (log ε): 234 (3.87), 273 sh (3.49); IR νmax: 3263, 2966, 2926, 2854, 1659, 1557, 1417 cm−1; 1H and 13C NMR data, see Table 3; HRESITOFMS (m/z): [M – H]– calcd for C16H24NO4, 294.1711; found, 294.1704.
Allostreptopyrrole E (5): greenish yellow amorphous solid; UV (MeOH) λmax, nm (log ε): 235 (3.90), 273 sh (3.49); IR νmax: 3270, 2964, 2927, 2858, 1659, 1555, 1416 cm−1; 1H and 13C NMR data, see Table 3; HRESITOFMS (m/z): [M + Na]+ calcd for C17H27NO4Na, 332.1832; found, 332.1838.
Methylation of 1
Allostreptopyrrole A (1, 2.0 mg, 0.007 mmol) and K2CO3 (4.4 mg, 0.032 mmol) were stirred in dry DMF (0.5 mL) at 50 °C for 10 min. Methyl iodide (19 μL, 0.32 mmol) was added and the mixture was stirred at this temperature for 12 h [29]. Reaction completion was monitored by TLC. The solution was diluted with water and extracted with EtOAc three times. The organic layer was washed with brine and evaporated to dryness to afford bismethylated derivative of 1 (1a, 1.9 mg, 88% yield).
DFT methodology
Prior to the calculations of the molecular properties of compounds 2 and 3, their conformational ensembles, datasets of a structure and population of each conformer [25], were determined using a Spartan 24 parallel package conformational search tool (Wavefunction Inc, USA), followed by geometry optimization of the most weighted conformers (Boltzmann distribution weight lower to 0.004). Eleven and seven weighted conformers for compounds 2 and 3 were obtained, respectively, each forming equilibrium mixtures. The quantum chemical method for the calculation of conformer distribution was ωB97X-V/6-311+G(2df,2p)[6-311G*] [30]. The CPCM solvation model for methanol was used [31]. Range-separated hybrid GGA (RSH-GGA) functional, including dispersive interaction with 6-31G* as the polarization basis set (ωB97X-D/6-31G* method), was used for energy and geometry optimization [32].
Bioactivity
Cytotoxicity and tyrosinase assays were carried out according to the procedures previously described [33,34]. The detailed procedures are available in Supporting Information File 1.
Supporting Information
Supporting Information File 1:
1D and 2D NMR, MS, UV, and IR spectra of compounds 1–5; experimental section including general experimental procedures, microorganism, detailed procedures for fermentation, extraction, isolation, and bioassays.
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
Tsuchimoto, T. Chem. – Eur. J.2011,17, 4064–4075. doi:10.1002/chem.201002248
Return to citation in text:
[1]
[2]
[3]
Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep.2006,23, 517–531. doi:10.1039/b605245m
Return to citation in text:
[1]
[2]
[3]
[4]
Xue, D.-Q.; Liu, H.-L.; Chen, S.-H.; Mollo, E.; Gavagnin, M.; Li, J.; Li, X.-W.; Guo, Y.-W. Chin. Chem. Lett.2017,28, 1190–1193. doi:10.1016/j.cclet.2017.03.040
Return to citation in text:
[1]
Cimino, G.; de Stefano, S.; Minale, L. Experientia1975,31, 1387–1389. doi:10.1007/bf01923200
Return to citation in text:
[1]
Stierle, D. B.; Faulkner, D. J. J. Org. Chem.1980,45, 4980–4982. doi:10.1021/jo01312a033
Return to citation in text:
[1]
El-Demerdash, A.; Tammam, M. A.; Atanasov, A. G.; Hooper, J. N. A.; Al-Mourabit, A.; Kijjoa, A. Mar. Drugs2018,16, 214. doi:10.3390/md16060214
Return to citation in text:
[1]
Kato, S.; Shindo, K.; Kawai, H.; Odagawa, A.; Matsuoka, M.; Mochizuki, J. J. Antibiot.1993,46, 892–899. doi:10.7164/antibiotics.46.892
Return to citation in text:
[1]
[2]
[3]
[4]
Han, X.; Liu, Z.; Zhang, Z.; Zhang, X.; Zhu, T.; Gu, Q.; Li, W.; Che, Q.; Li, D. J. Nat. Prod.2017,80, 1684–1687. doi:10.1021/acs.jnatprod.7b00016
Return to citation in text:
[1]
[2]
Kwon, H. C.; Espindola, A. P. D. M.; Park, J.-S.; Prieto-Davó, A.; Rose, M.; Jensen, P. R.; Fenical, W. J. Nat. Prod.2010,73, 2047–2052. doi:10.1021/np1006229
Return to citation in text:
[1]
[2]
Raju, R.; Piggott, A. M.; Barrientos Diaz, L. X.; Khalil, Z.; Capon, R. J. Org. Lett.2010,12, 5158–5161. doi:10.1021/ol102162d
Return to citation in text:
[1]
[2]
[3]
[4]
Stankovic, N.; Senerovic, L.; Ilic-Tomic, T.; Vasiljevic, B.; Nikodinovic-Runic, J. Appl. Microbiol. Biotechnol.2014,98, 3841–3858. doi:10.1007/s00253-014-5590-1
Return to citation in text:
[1]
[2]
Liu, D.-Z.; Liang, B.-W. Magn. Reson. Chem.2014,52, 57–59. doi:10.1002/mrc.4031
Return to citation in text:
[1]
Liu, D.-Z.; Liang, B.-W.; Li, X.-F. Chem. Biodiversity2015,12, 153–156. doi:10.1002/cbdv.201400001
Return to citation in text:
[1]
Liu, D.-Z.; Liang, B.-W. J. Antibiot.2014,67, 415–417. doi:10.1038/ja.2014.8
Return to citation in text:
[1]
Clark, B. R.; O'Connor, S.; Fox, D.; Leroy, J.; Murphy, C. D. Org. Biomol. Chem.2011,9, 6306–6311. doi:10.1039/c1ob05667k
Return to citation in text:
[1]
Huang, M.-J.; Rao, M. P. N.; Salam, N.; Xiao, M.; Huang, H.-Q.; Li, W.-J. Int. J. Syst. Evol. Microbiol.2017,67, 288–293. doi:10.1099/ijsem.0.001617
Return to citation in text:
[1]
[2]
Sahu, A. K.; Quadri, S. R.; Agasar, D.; Ruwaili, J. A.; Jun-Li, W.; Dastager, S. G. J. Antibiot.2017,70, 1000–1003. doi:10.1038/ja.2017.82
Return to citation in text:
[1]
Suga, T.; Kimura, T.; Inahashi, Y.; Iwatsuki, M.; Nonaka, K.; Také, A.; Matsumoto, A.; Takahashi, Y.; Ōmura, S.; Nakashima, T. J. Antibiot.2018,71, 619–625. doi:10.1038/s41429-018-0055-x
Return to citation in text:
[1]
Elsbaey, M.; Samaru, Y.; Elekhnawy, E.; Oku, N.; Igarashi, Y. J. Antibiot.2024,77, 393–396. doi:10.1038/s41429-024-00705-7
Return to citation in text:
[1]
[2]
[3]
Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Nat. Prod. Rep.2019,36, 1005–1030. doi:10.1039/c9np00018f
Return to citation in text:
[1]
[2]
Lu, Q.; Zhang, L.; He, G.-R.; Liang, H.-X.; Du, G.-H.; Cheng, Y.-X. Chem. Biodiversity2007,4, 2948–2952. doi:10.1002/cbdv.200790244
Return to citation in text:
[1]
Ortega, M. J.; Zubia, E.; Carballo, J. L.; Salvá, J. Tetrahedron1997,53, 331–340. doi:10.1016/s0040-4020(96)00989-1
Return to citation in text:
[1]
Sutherland, H. S.; Tong, A. S. T.; Choi, P. J.; Blaser, A.; Conole, D.; Franzblau, S. G.; Lotlikar, M. U.; Cooper, C. B.; Upton, A. M.; Denny, W. A.; Palmer, B. D. Bioorg. Med. Chem.2019,27, 1292–1307. doi:10.1016/j.bmc.2019.02.026
Return to citation in text:
[1]
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys.1980,72, 650–654. doi:10.1063/1.438955
Return to citation in text:
[1]
Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. J. Comput. Chem.2003,24, 669–681. doi:10.1002/jcc.10189
Return to citation in text:
[1]
Chai, J.-D.; Head-Gordon, M. J. Chem. Phys.2008,128, 084106. doi:10.1063/1.2834918
Return to citation in text:
[1]
Flores Hernandez, F. Y.; Khandual, S.; Ramírez López, I. G. Asian Pac. J. Trop. Biomed.2017,7, 14–19. doi:10.1016/j.apjtb.2016.10.011
Return to citation in text:
[1]
Chiari, M. E.; Joray, M. B.; Ruiz, G.; Palacios, S. M.; Carpinella, M. C. Food Chem.2010,120, 10–14. doi:10.1016/j.foodchem.2009.09.061
Return to citation in text:
[1]
Kwon, H. C.; Espindola, A. P. D. M.; Park, J.-S.; Prieto-Davó, A.; Rose, M.; Jensen, P. R.; Fenical, W. J. Nat. Prod.2010,73, 2047–2052. doi:10.1021/np1006229
Kato, S.; Shindo, K.; Kawai, H.; Odagawa, A.; Matsuoka, M.; Mochizuki, J. J. Antibiot.1993,46, 892–899. doi:10.7164/antibiotics.46.892
13.
Han, X.; Liu, Z.; Zhang, Z.; Zhang, X.; Zhu, T.; Gu, Q.; Li, W.; Che, Q.; Li, D. J. Nat. Prod.2017,80, 1684–1687. doi:10.1021/acs.jnatprod.7b00016
14.
Kwon, H. C.; Espindola, A. P. D. M.; Park, J.-S.; Prieto-Davó, A.; Rose, M.; Jensen, P. R.; Fenical, W. J. Nat. Prod.2010,73, 2047–2052. doi:10.1021/np1006229
15.
Raju, R.; Piggott, A. M.; Barrientos Diaz, L. X.; Khalil, Z.; Capon, R. J. Org. Lett.2010,12, 5158–5161. doi:10.1021/ol102162d
Sutherland, H. S.; Tong, A. S. T.; Choi, P. J.; Blaser, A.; Conole, D.; Franzblau, S. G.; Lotlikar, M. U.; Cooper, C. B.; Upton, A. M.; Denny, W. A.; Palmer, B. D. Bioorg. Med. Chem.2019,27, 1292–1307. doi:10.1016/j.bmc.2019.02.026



![[1860-5397-20-174-1]](/bjoc/content/figures/1860-5397-20-174-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-20-174-2]](/bjoc/content/figures/1860-5397-20-174-2.png?scale=2.2&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-174-i1.svg?max-width=637&scale=1.18182) +15 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.87), 273 sh (3.49); IR (ATR) νmax: 3263, 2964, 2925, 2854, 1658, 1556, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1554.
+15 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.87), 273 sh (3.49); IR (ATR) νmax: 3263, 2964, 2925, 2854, 1658, 1556, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1554.![[Graphic 2]](/bjoc/content/inline/1860-5397-20-174-i2.svg?max-width=637&scale=1.18182) −6.1 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.82), 276 sh (3.46); IR (ATR) νmax: 3265, 2925, 2856, 1657, 1555, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1556.
−6.1 (c 0.10, MeOH); UV (MeOH) λmax, nm (log ε): 235 (3.82), 276 sh (3.46); IR (ATR) νmax: 3265, 2925, 2856, 1657, 1555, 1417 cm−1; 1H and 13C NMR data, see Table 2; HRESITOFMS (m/z): [M – H]– calcd for C15H22NO4, 280.1554; found, 280.1556.