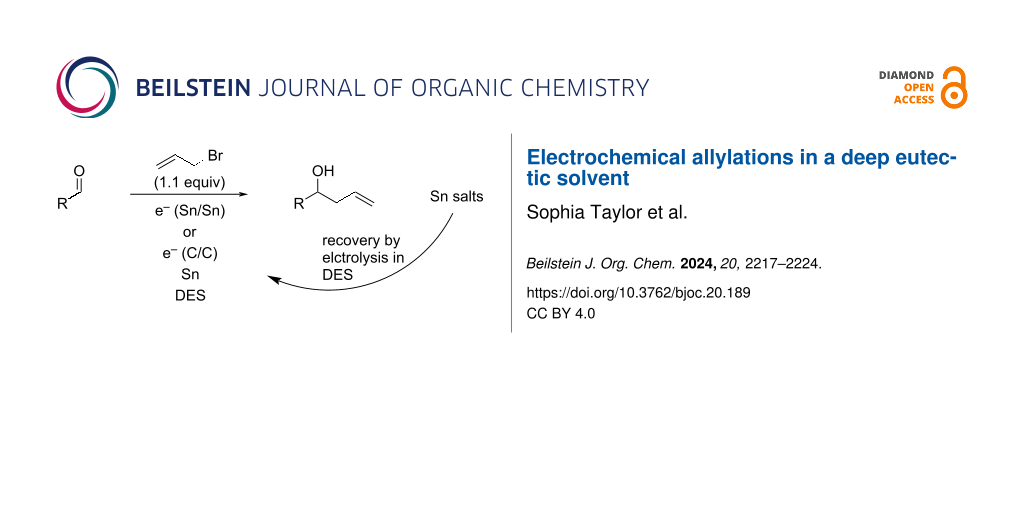
Abstract
Electrosynthesis is a technique that is attracting increased attention and has many appealing features, particularly its potential greenness. At the same time, electrosynthesis requires a solvent and a supporting electrolyte in order for current to pass through the reaction. These are effectively consumable reagents unless a convenient means of recycling can be developed. As part of our interest in unusual solvents and electrochemistry, we explored the application of simple, inexpensive, and recyclable deep eutectic solvents to the allylation of carbonyls. While several sets of conditions were developed, the goal of avoiding stoichiometric amounts of metal has proven elusive. Still, a deep eutectic solvent can be used to plate out and thus recover the metal used, offering an interesting new option for electrochemical allylations.

Graphical Abstract
Introduction
The last several years have witnessed a tremendous resurgence of interest in electrochemistry in the area of organic synthesis [1]. While there are many reasons for this renewed interest, two major motivations are the unmatched control of oxidation or reduction potential that can be achieved and the environmentally friendly aspect of having electrons as the only consumed reagent. This latter reason is certainly an advantage in many cases, but its effective realization is limited by the need for both a solvent as well as a supporting electrolyte to allow for the flow of current through the reaction. Although some imaginative options have been reported, they tend to be quite limited in scope. Room temperature ionic liquids (RTILs) were viewed as interesting options that would combine both the solvent and the electrolyte into one component and could be readily recycled due to their non-volatile nature [2-15]. Further, many of these species featured very wide electrochemical windows. In practice, however, RTILs are expensive compared to conventional solvents. Most of them are also quite viscous, which severely limits their use in synthetic electrochemistry [16].
These same expense and viscosity issues plague the application of RTILs in any area. Driven by this limitation, deep eutectic solvents (DES) were reported and have been heavily promoted as cost-effective replacements for RTILs in many applications [17,18]. One of the early and very effective applications of DES was in the area of electroplating as well as metal recovery by electrodeposition [19]. Despite this fascinating potential, very little has been reported in terms of their use in electrosynthesis [20-24].
Given our interest in both electrosynthesis and DES, we opted to explore this combination in the area of electrochemical allylation. The allylation of carbonyls is a valuable reaction that has been explored under a wide range of conditions, including several reports using electrochemical conditions [25,26]. Recently, an allylation in DES has also been reported, but using indium as the reducing metal under standard Barbier-type conditions, but not electrochemical ones [27]. While the unusual DES (acetylcholine chloride/acetamide 2:1 molar ratio with added ammonium chloride) could be recycled five times with a modest decrease in yield (99% to 65%), the requirement of using suprastoichiometric amounts of expensive indium metal for each reaction is a significant drawback. The potential to avoid this limitation by using electrochemistry with catalytic amounts of metal or perhaps by recovering and recycling the metal with a recyclable solvent was the goal motivating this project. With this as a background, we undertook an investigation of electrochemical allylation in DES.
Results and Discussion
While there are many potential DES that are known, one that would be stable to highly reducing conditions was desired for the allylation reaction. Further, to make the reaction conditions similar to standard electrochemical ones, a solvent with a tetraalkylammonium salt, the first DES that was studied was the 1:3 molar ratio of tetrabutylammonium bromide and ethylene glycol (TBAB/EG) [28]. Using the reaction of p-anisaldehyde with allyl bromide as a test case, reactions were performed using three sets of different sacrificial electrodes as well as non-sacrificial graphite. As can be seen in Table 1, tin (entry 1) resulted in good conversion to the allylation product, while zinc, magnesium, and graphite (Table 1, entries 3–5) displayed no reaction at all. This observation was somewhat surprising considering that both zinc and tin are very commonly used in conventional allylations in addition to as electrode materials for electrochemical allylations [29-48]. Further optimization employing tin (Table 1, entry 2) enabled near complete conversion to the desired product by using 2.5 F/mol of current passed through the solution at a constant current of 20 mA.
Table 1: Influence of electrode material.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-189-i1.svg?max-width=637&scale=1.0)
|
|||
| Entry | Electrode | F/mol | % Conversion |
| 1 | Sn/Sn | 2 | 86 |
| 2 | Sn/Sn | 2.5 | 91a |
| 3 | Zn/Zn | 2.5 | 0 |
| 4 | Mg/Mg | 2.5 | 0 |
| 5 | C/C | 2.5 | 0 |
aIsolated yield of product 78%.
With this promising start, several other aldehydes and two ketones were explored under the same reaction conditions (Table 2). A range of aromatic aldehydes worked well as well as one aliphatic aldehyde (Table 2, entry 13) and one alkenyl aldehyde (Table 2, entry 10), although the highly electron-rich dimethylaminobenzaldehyde (Table 2, entry 4) afforded only recovered starting material. Both ketones (Table 2, entries 11 and 12) failed to react and resulted in just recovered starting material. 4-Nitrobenzaldehyde (Table 2, entry 5) also failed to afford any allylation product, although in this case, reduction of the nitro group to an amino group was observed and the resulting 4-aminobenzaldehyde is likely too electron-rich to undergo allylation.
Table 2: Aldehyde variations.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-189-i2.svg?max-width=637&scale=1.0)
|
||
| Entry | R | Isolated yield |
| 1 | p-MeOC6H4 | 78% |
| 2 | p-BrC6H4 | 82% |
| 3 | p-CNC6H4 | 80% |
| 4 | p-Me2NC6H4 | NR |
| 5 | p-NO2C6H4 | 0%a |
| 6 | p-MeC6H4 | 85% |
| 7 | m-MeC6H4 | 79% |
| 8 | o-MeC6H4 | 76% |
| 9 | Ph | 68% |
| 10 | cinnamyl | 77% |
| 11 | acetophenone | NR |
| 12 | cyclohexanone | NR |
| 13 | C6H11 | 84% |
aOnly nitro group reduced, no allylation.
The use of other halides was also explored (Table 3). Switching to allyl chloride (Table 3, entry 2) did result in partial conversion, but the reaction was much less efficient than for allyl bromide. More substituted allyl bromides, such as crotyl and prenyl bromide (Table 3, entries 3 and 4) did react, although they afforded only partial conversion when using 2.5 F/mol of current. In terms of regiochemistry, addition at the more substituted end (gamma addition) was the major product in both cases, although alpha addition was also observed. In the case of crotyl bromide, no diastereoselectivity was noted for the gamma product. Non-allylic halides such as benzyl bromide, propargyl bromide, and ethyl bromoacetate (Table 3, entries 5–7) did not result in carbonyl addition. The benzyl bromide was recovered intact, while for the other two, they are both sufficiently volatile that they would have been lost during the work-up. For all three, the aldehyde was recovered unreacted.
Table 3: Halide variations.
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-189-i3.svg?max-width=637&scale=1.0)
|
||
| Entry | R–X | % Conversiona |
| 1 | allyl bromide | 100% |
| 2 | allyl chloride | 50% |
| 3 | crotyl bromide | 85%b |
| 4 | prenyl bromide | 45%c |
| 5 | ethyl bromoacetate | NR |
| 6 | benzyl bromide | NR |
| 7 | propargyl bromide | NR |
aDetermined by 1H NMR of the crude extract; bγ to α addition 2.2:1.0, 1:1 diastereomeric mixture of the γ product; cγ to α addition 3:1.
While the use of DES as solvent and electrolyte for electrochemical allylation had been demonstrated, there was no particular advantage to this method over non-DES options without recycling of the DES. To explore this aspect, the allylation of anisaldehyde with allyl bromide was undertaken using TBAB/EG. As can be seen in Table 4, the DES could be recycled two times before incomplete conversion was noted. A more significant problem was the steady loss of DES during the product extraction stage. Using 3 mL of methoxycyclopentane in a single extraction, the amount of DES steadily decreased by roughly 0.5 mL for each recycling. This is likely due to partial solubility of ethylene glycol in methoxycyclopentane as ethylene glycol could be clearly seen in the 1H NMR spectrum of the crude reaction extracts. It should also be noted that the lower isolated yields most likely reflect mechanical losses during extraction and chromatographic separation as the crude spectra do not show significant amounts of side products.
Table 4: Recycling for the allylation of p-anisaldehyde in TBAB/EG DES and Sn electrodes.
| Cycle | Volume of DES recovered | Conversion/isolated yielda |
| 1 | 2.5 mL | 100%/78% |
| 2 | 2.0 mL | 100%/77% |
| 3 | 1.5 mL | 100%/79% |
| 4 | 1.25 mL | 80%/60% |
aDetermined by 1H NMR of the crude extract.
Another problem that was noted in these recycling experiments was a build-up of tin salt byproducts that made the DES more and more viscous in each recycling. Avoiding the use of a sacrificial electrode could help this aspect as well as reducing the waste generated in these allylations. While non-sacrificial graphite electrodes had failed to result in any reaction, it seemed possible that the use of a catalytic amount of tin metal or a tin salt with graphite electrodes would result in a superior reaction due to in situ reduction and/or activation of the tin. This variation was explored as seen in Table 5. Use of 0.5 equivalents of tin metal (Table 5, entry 1) did result in partial conversion to product, which increased to complete conversion when 1.5 equivalents of tin were used (Table 5, entry 3). Upon attempted recycling of this reaction, very little conversion was noted, indicating that this method simply exchanged one form of consumed tin (the electrode) for another (the metal powder). We also explored the use of SnCl2 as the tin source. Little to no reaction occurred under a variety of constant current conditions (Table 5, entries 5 and 6), but using two equivalents at a constant potential of 2.0 V (Table 5, entry 8) resulted in complete conversion to the allylation product. As before, attempts to directly recycle the salt/DES mixture failed to afford any allylation.
Table 5: Tin metal with non-sacrificial electrodes.
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-189-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Tin source | Equivalents of tin | % Conversiona |
| 1 | Sn powder | 0.5 | 15% |
| 2 | Sn powder | 1 | 40% |
| 3 | Sn powder | 1.5 | 100% |
| 4 | SnCl2 | 1 | 10% |
| 5 | SnCl2 | 2b | NR |
| 6 | SnCl2 | 2c | NR |
| 7 | SnCl2 | 2d | 50% |
| 8 | SnCl2 | 2e | 100% |
aDetermined by 1H NMR of the crude extract; b100 mA constant current; c50 mA constant current; d100 mA constant current and 10% by volume water added; econstant potential of 2 V.
Next, it was decided to compare the results of the sacrificial tin electrodes at a constant current of 20 mA with the non-sacrificial glassy carbon electrodes and tin(II) chloride at a constant potential of 2 V for a set of aldehydes. As can be seen in Table 6, three of the four cases (entries 1, 2, and 4), the glassy carbon conditions afforded better yields of the allylation product. Only in the case of the cyclohexanecarboxaldehyde (Table 6, entry 3) was the yield lower and in this case this lower yield was the result of incomplete conversion of the aldehyde.
Table 6: Electrode comparison in TBAB/EG.
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-189-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Aldehyde | Sn/Sn isolated yield | C/C with SnCl2 isolated yield |
| 1 | p-anisaldehyde | 83% | 91% |
| 2 | p-tolualdehyde | 77% | 83% |
| 3 |
cyclohexane-
carboxaldehyde |
68% | 58%a |
| 4 | cinnamaldehyde | 61% | 85% |
a12% recovered aldehyde.
For as successful as the TBAB/EG conditions were, TBAB is not inexpensive. Choline chloride is much less expensive and is particularly well known and frequently used in DESs. It is known that choline chloride and ethylene glycol will also form a DES when combined in a 1:2 molar ratio [49]. This solvent is much more viscous than the TBAB/EG one, and an initial attempt to use the same 20 mA constant current conditions with sacrificial tin electrodes resulted in 50% conversion to the allylation product and starting material after passing 2.5 F/mol of current. This poor conversion could be significantly improved by adding 10 volume percent of water to the reaction mixture, which also visibly decreased the viscosity. While complete conversion was still not achieved (67% conversion), it was a considerable improvement. Using glassy carbon electrodes with two equivalents of tin(II) chloride under a constant potential of 2 V, but still in CC/EG with 10 volume percent water, resulted in further improvement (75% conversion) for the same reaction with anisaldehyde. Comparing these two variations always resulted in improved conversions and yields for the glassy carbon conditions, although the results were generally not superior to those obtained in TBAB/EG (Table 7). Interestingly cyclohexanecarboxaldehyde (Table 7, entry 3) was again the outlier as it afforded an excellent yield and complete conversion in the case of glassy carbon in CC/EG.
Table 7: Electrode comparison in CC/EG.
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-189-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Aldehyde | Sn/Sn isolated yield | C/C with SnCl2 isolated yield |
| 1 | p-anisaldehyde | 34%a | 62%b |
| 2 | p-tolualdehyde | 24%b | 67%c |
| 3 |
cyclohexane-
carboxaldehyde |
52%c | 96% |
| 4 | cinnamaldehyde | 58%d | 62%b |
aReaction 50% completion; breaction 75% completion; creaction 67% completion; dreaction 80% completion.
While the CC/EG system did not appear in general to be as successful as the TBAB/EG one in terms of yields and conversions, it was decided to explore DES recycling (Table 8). As with TBAB/EG, the allylation of p-anisaldehyde with allyl bromide was conducted using 2 equivalents of tin(II) chloride in CC/EG with 10 volume percent water at a constant potential of 2 V until 2.5 F/mol of current had been passed through the reaction. The product and unreacted starting material were recovered via extraction with methoxycyclopentane and the CC/EG used in another cycle. Through two recyclings, the reactions were similar in efficiency, although the DES became increasingly viscous as the tin byproducts built up and it became impractical to recycle the DES further.
![[Graphic 7]](/bjoc/content/inline/1860-5397-20-189-i7.svg?max-width=637&scale=1.0)
While the DES clearly had some ability to be recycled, the use of catalytic amounts of metal had uniformly failed. Recognizing that DES have been used extensively for electroplating and metal deposition, though it seemed that this might provide an opportunity for metal recovery and reuse [19]. To this end, the DES mixture from cycle 3 of Table 8 following product extraction was electrolyzed at 100 mA constant current using glassy carbon electrodes until 2 F/mol of current had passed. This deposited a metal clump on the electrode that was removed and then analyzed using X-ray fluorescent spectroscopy to determine that the deposited metal was tin, with traces of other metals consistent with the purity of the initial tin source. The tin recovery was 99% of the theoretical. To explore recycling of this recovered tin, 1 mmol was used in a 0.5 mmol scale allylation of anisaldehyde under the constant potential conditions with glassy carbon electrodes and fresh DES. This reaction afforded the anticipated product in 74% yield, thus demonstrating that the tin can be recovered and recycled.
Conclusion
In conclusion, we have demonstrated the ability to use DES as a combined solvent and electrolyte for electrosynthesis. It can be recycled, although product extraction does result in a slow but detectible loss of the ethylene glycol. Attempts to use catalytic amounts of metal with non-sacrificial electrodes in place of the sacrificial tin electrodes were not successful, but we were able to demonstrate efficient and near quantitative recovery of the metal by electrolysis after product extraction and this recovered metal can be used again for further allylations. Further efforts to improve the efficiency and enable catalytic metal use are underway.
Experimental
Preparation of deep eutectic solvents
Tetrabutylammonium bromide/ethylene glycol (1:3 molar ratio) deep eutectic solvent
To 8.0 grams of tetrabutylammonium bromide (TBAB) were added 4.7 grams of ethylene glycol (EG). The resulting mixture was heated to 70 °C until a homogeneous liquid formed. It was stored at this same temperature between uses.
Choline chloride/ethylene glycol (1:2 molar ratio) deep eutectic solvent
To 6.98 grams of choline chloride (CC) were added 6.2 grams of EG and the resulting mixture was heated to 70 °C until a homogeneous liquid formed. It was stored at this same temperature between uses.
General Sn/Sn procedure with TBAB/EG
To a 10 mL ElectraSyn 2.0 vial containing a magnetic stir bar, the carbonyl compound (0.5 mmol), allyl bromide (0.6 mmol), and TBAB/EG DES (2 mL) were added. Tin electrodes were used as the working and counter electrodes and were submerged into the reaction. The reaction was performed under constant current conditions of 20 mA with no reference electrode until 2.5 F/mol was passed. The current was programmed to alternate every five minutes to minimize any potential fouling of the electrode surface. Following completion of each reaction, the mixture was transferred to a separatory funnel with the aid of 10 mL of deionized (DI) water followed by 20 mL of diethyl ether. The organic layer was separated, dried with anhydrous magnesium sulfate, filtered, and the solvent removed in vacuo to afford the crude product, which was first analyzed by 1H NMR spectroscopy and then purified using flash column chromatography. In between reactions, the tin electrodes were rinsed with DI water and acetone, then polished using diamond polish. This helped prevent buildup on the electrode surfaces.
General C/C procedure with TBAB/EG
To a 10 mL ElectraSyn 2.0 vial containing a magnetic stir bar were added the carbonyl compound (0.5 mmol), allyl bromide (0.6 mmol), and TBAB/EG DES (2 mL) along with SnCl2 (0.1896 g, 1 mmol). Graphite electrodes were used as the working and counter electrodes and were submerged into the reaction. The ElectraSyn was programmed to run the reaction under constant potential conditions of 2 V with no reference electrode until 2.5 F/mol was passed. Following completion of each reaction, the mixture was transferred to a separatory funnel with the aid of 10 mL of DI water followed by 20 mL of diethyl ether. The organic layer was separated, dried with anhydrous magnesium sulfate, filtered, and the solvent removed in vacuo to afford the crude product, which was first analyzed by 1H NMR spectroscopy and then purified using flash column chromatography. The graphite electrodes were not polished in between reactions, but were rinsed with DI water and then acetone.
General Sn/Sn procedure with CC/EG
To a 10 mL ElectraSyn 2.0 vial containing a magnetic stir bar, the carbonyl compound (0.5 mmol), allyl bromide (0.6 mmol), CC/EG DES (2 mL), and 10% DI water (0.2 mL) were added. Tin electrodes were used as the working and counter electrodes and were submerged into the reaction. The ElectraSyn was programmed to run the reaction under constant current conditions of 20 mA with no reference electrode until 2.5 F/mol was passed. The current was programmed to alternate every five minutes. Following completion of each reaction, the mixture was transferred to a separatory funnel with the aid of 10 mL of DI water followed by 20 mL of diethyl ether. The organic layer was separated, dried with anhydrous magnesium sulfate, filtered, and the solvent removed in vacuo to afford the crude product, which was first analyzed by 1H NMR spectroscopy and then purified using flash column chromatography. The electrodes were washed with DI water and acetone before polishing.
General C/C procedure with CC/EG
To a 10 mL ElectraSyn 2.0 vial containing a magnetic stir bar were added the carbonyl compound (0.5 mmol), allyl bromide (0.6 mmol), CC/EG DES (2 mL), and 10% DI water (0.2 mL) along with SnCl2 (0.1896 g, 1 mmol). Graphite electrodes were used as the working and counter electrodes and were submerged into the reaction. The ElectraSyn was programmed to run the reaction under constant potential conditions of 2 V with no reference electrode until 2.5 F/mol was passed. Following completion of each reaction, the mixture was transferred to a separatory funnel with the aid of 10 mL of DI water followed by 20 mL of diethyl ether. The organic layer was separated, dried with anhydrous magnesium sulfate, filtered, and the solvent removed in vacuo to afford the crude product, which was first analyzed by 1H NMR spectroscopy and then purified using flash column chromatography. The graphite electrodes were not polished in between reactions, but were rinsed with DI water and then acetone.
DES and tin metal recycling
For the recyclability trials, the electrolysis vial was opened and about 3 mL of methoxycyclopentane was pipetted directly onto the reaction solution. Then, the vial was capped with a rubber stopper. The methoxycyclopentane acted similarly to the diethyl ether in previous separatory methods by extracting the product from the DES. After letting the methyoxycyclopentane and DES layers separate, the methoxycyclopentane layer was removed using a pipette, placed into a round-bottomed flask, and concentrated in vacuo. This remaining DES could be used directly in subsequent reactions.
If SnCl2 was used in the reaction, electrolysis of the tin metal could be achieved after workup with methoxycyclopentane. Once all product was extracted, the graphite electrodes were submerged in the remaining DES solution in the vial and the tin metal (around 3 mmol) was electrolyzed at 100 mA constant current until 2 F/mol had passed. The resulting metal clump on the electrode was removed using a scoopula for further use and analysis using a Thermo Scientific Niton XL3t X-Ray Fluorescent Spectroscopy analyzer.
Product characterization
1-(4-Methoxyphenyl)-3-buten-1-ol [39]: 1H NMR (300 MHz, CDCl3) 7.27 (d, J = 8.58, 2H), 6.86 (d, J = 5.82, 2H), 5.83–5.73 (m, 1H), 5.17–5.10 (m, 2H), 4.67 (t, J = 6.18, 1H), 3.80 (s, 3H), 2.49 (t, J = 4.8, 2H).
1-(4-Bromophenyl)-3-buten-1-ol [50]: 1H NMR (300 MHz, CDCl3) 7.43 (d, J = 8.58, 2H), 7.18 (d, J = 8.25, 2H), 5.80–5.65 (m, 1H), 5.13–5.07 (m, 2H), 4.64 (t, J = 6.51, 1H), 2.43 (t, J = 7.2, 2H).
4-(1-Hydroxybut-3-enyl)benzonitrile [51]: 1H NMR (500 MHz, CDCl3) 7.59 (d, J = 8.6, 2H), 7.44 (d, J = 7.45, 2H), 5.78–5.70 (m, 1H), 5.14–5.10 (m, 2H), 4.76 (t, J = 2.85, 1H), 2.48–2.41 (m, 2H).
1-Cyclohexyl-3-buten-1-ol [39]: 1H NMR (300 MHz, CDCl3) 6.0–5.7 (m, 1H), 5.2–5.0 (m, 1H), 3.5–3.3 (m, 1H), 2.4–2.2 (m, 1H), 2.1–2.0 (m, 1H), 1.9–1.6 (m, 4H), 1.5–1.0 (m, 4H).
1-(4-Methylphenyl)-3-buten-1-ol [39]: 1H NMR (300 MHz, CDCl3) 7.24 (d, J = 8.25, 2H), 7.16 (d, J = 8.22, 2H), 5.87–5.73 (m, 1H), 5.17–5.10 (m, 2H), 4.89 (s, 1H), 4.69 (t, J = 6.54, 1H), 2.51 (t, J = 6.6, 2H), 2.35 (s, 3H).
1-(3-Methylphenyl)-3-buten-1-ol [50]: 1H NMR (500 MHz, CDCl3) 7.24 (t, J = 7.45, 1H), 7.18 (s, 1H), 7.14 (d, J = 8.05, 1H), 7.09 (d, J = 7.45, 1H), 5.85–5.77 (m, 1H), 5.18–5.12 (m, 2H), 4.69 (t, J = 5.15, 1H), 2.50 (t, J = 7.45, 2H), 2.36 (s, 3H).
1-(2-Methylphenyl)-3-buten-1-ol [50]: 1H NMR (300 MHz, CDCl3) 7.27–7.14 (m, 4H), 5.91–5.80 (m, 1H), 5.22–5.14 (m, 2H), 4.97 (t, J = 3.6, 1H), 2.49 (t, J = 8.25, 2H), 2.34 (s, 3H).
1-Phenyl-3-buten-1-ol [39]: 1H NMR (300 MHz, CDCl3) 7.38–7.35 (m, 3H), 5.85–5.76 (m, 2H), 5.15–5.12 (m, 2H), 4.73 (t, J = 1.38, 1H), 2.51 (t, J = 1.02, 2H).
1-Phenyl-1,5-hexadien-3-ol [52]: 1H NMR (300 MHz, CDCl3) 7.41–7.22 (m, 5H), 6.61 (d, J = 16.5, 1H), 6.25 (dd, = 6.54, 15.78 Hz, 1H), 5.90–5.79 (m, 1H), 5.22–5.15 (m, 2H), 4.36 (q, J = 5.85, 1H), 2.41 (q, J = 8.94, 2H).
Data Availability Statement
The data that supports the findings of this study is available from the corresponding author upon reasonable request.
References
-
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Nozaki, S.; Suzuki, Y.; Goto, T. Electrochim. Acta 2024, 493, 144431. doi:10.1016/j.electacta.2024.144431
Return to citation in text: [1] -
Kong, X.; Liu, Q.; Chen, Y.; Wang, W.; Chen, H.-F.; Wang, W.; Zhang, S.; Chen, X.; Cao, Z.-Y. Green Chem. 2024, 26, 3435–3440. doi:10.1039/d3gc04528e
Return to citation in text: [1] -
David, M.; Galli, E.; Brown, R. C. D.; Feroci, M.; Vetica, F.; Bortolami, M. Beilstein J. Org. Chem. 2023, 19, 1966–1981. doi:10.3762/bjoc.19.147
Return to citation in text: [1] -
Witherspoon, E.; Ling, P.; Winchester, W.; Zhao, Q.; Ibrahim, A.; Riley, K. E.; Wang, Z. ACS Omega 2022, 7, 42828–42834. doi:10.1021/acsomega.2c04748
Return to citation in text: [1] -
Rocco, D.; Chiarotto, I.; Mattiello, L.; Pandolfi, F.; Zane, D.; Feroci, M. Pure Appl. Chem. 2019, 91, 1709–1715. doi:10.1515/pac-2018-1118
Return to citation in text: [1] -
Yimin, D.; Lanli, N.; Hui, L.; Jiaqi, Z.; Linping, Y.; Qiuju, F. Int. J. Electrochem. Sci. 2018, 13, 1084–1095. doi:10.20964/2018.01.85
Return to citation in text: [1] -
Kathiresan, M.; Velayutham, D. Chem. Commun. 2015, 51, 17499–17516. doi:10.1039/c5cc06961k
Return to citation in text: [1] -
Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Phys. Chem. Chem. Phys. 2015, 17, 19247–19254. doi:10.1039/c5cp00095e
Return to citation in text: [1] -
Kronenwetter, H.; Husek, J.; Etz, B.; Jones, A.; Manchanayakage, R. Green Chem. 2014, 16, 1489–1495. doi:10.1039/c3gc41641k
Return to citation in text: [1] -
Orsini, M.; Chiarotto, I.; Feeney, M. M. M.; Feroci, M.; Sotgiu, G.; Inesi, A. Electrochem. Commun. 2011, 13, 738–741. doi:10.1016/j.elecom.2011.04.025
Return to citation in text: [1] -
Liu, Y.-Z.; Lin, M.-Y.; Xiao, L.-P.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1168–1172. doi:10.1002/cjoc.200890214
Return to citation in text: [1] -
Mellah, M.; Zeitouny, J.; Gmouh, S.; Vaultier, M.; Jouikov, V. Electrochem. Commun. 2005, 7, 869–874. doi:10.1016/j.elecom.2005.06.002
Return to citation in text: [1] -
Doherty, A. P.; Brooks, C. A. Electrochim. Acta 2004, 49, 3821–3826. doi:10.1016/j.electacta.2003.12.058
Return to citation in text: [1] -
Mellah, M.; Gmouh, S.; Vaultier, M.; Jouikov, V. Electrochem. Commun. 2003, 5, 591–593. doi:10.1016/s1388-2481(03)00136-x
Return to citation in text: [1] -
Bornemann, S.; Handy, S. T. Molecules 2011, 16, 5963–5974. doi:10.3390/molecules16075963
Return to citation in text: [1] -
Smith, E. L.; Abbott, A. P.; Ryder, K. S. Chem. Rev. 2014, 114, 11060–11082. doi:10.1021/cr300162p
Return to citation in text: [1] -
Hansen, B. B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J. M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B. W.; Gurkan, B.; Maginn, E. J.; Ragauskas, A.; Dadmun, M.; Zawodzinski, T. A.; Baker, G. A.; Tuckerman, M. E.; Savinell, R. F.; Sangoro, J. R. Chem. Rev. 2021, 121, 1232–1285. doi:10.1021/acs.chemrev.0c00385
Return to citation in text: [1] -
Protsenko, V. Coatings 2024, 14, 375. doi:10.3390/coatings14040375
Return to citation in text: [1] [2] -
Mousa, M. O.; Adly, M. E.; Mahmoud, A. M.; El-Nassan, H. B. ACS Omega 2024, 9, 14198–14209. doi:10.1021/acsomega.3c09790
Return to citation in text: [1] -
El-Nassan, H. B.; El-Mosallamy, S. S.; Mahmoud, A. M. Sustainable Chem. Pharm. 2023, 35, 101207. doi:10.1016/j.scp.2023.101207
Return to citation in text: [1] -
Osman, E. O.; Mahmoud, A. M.; El-Mosallamy, S. S.; El-Nassan, H. B. J. Electroanal. Chem. 2022, 920, 116629. doi:10.1016/j.jelechem.2022.116629
Return to citation in text: [1] -
Trujillo, S. A.; Peña-Solórzano, D.; Bejarano, O. R.; Ochoa-Puentes, C. RSC Adv. 2020, 10, 40552–40561. doi:10.1039/d0ra06871c
Return to citation in text: [1] -
Golgovici, F.; Anicai, L.; Florea, A.; Visan, T. Curr. Nanosci. 2020, 16, 478–494. doi:10.2174/1573413715666190206145036
Return to citation in text: [1] -
Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207–2293. doi:10.1021/cr00022a010
Return to citation in text: [1] -
Denmark, S. E.; Fu, J. Chem. Rev. 2003, 103, 2763–2794. doi:10.1021/cr020050h
Return to citation in text: [1] -
Gonzalez-Gallardo, N.; Saavedra, B.; Guillena, G.; Ramón, D. J. Appl. Organomet. Chem. 2021, 35, e6418. doi:10.1002/aoc.6418
Return to citation in text: [1] -
Yusof, R.; Abdulmalek, E.; Sirat, K.; Rahman, M. B. A. Molecules 2014, 19, 8011–8026. doi:10.3390/molecules19068011
Return to citation in text: [1] -
Zhang, J.; Zhang, L.; Xie, W.; Chen, M.; Zhang, C.; Qin, Y.; Zhao, J.; Wang, F.; Liu, Z.-Q. Green Chem. 2024, 26, 7002–7006. doi:10.1039/d4gc01838a
Return to citation in text: [1] -
Zhang, Q.; Liang, K.; Guo, C. Angew. Chem., Int. Ed. 2022, 61, e202210632. doi:10.1002/anie.202210632
Return to citation in text: [1] -
Torabi, S.; Jamshidi, M.; Amooshahi, P.; Mehrdadian, M.; Khazalpour, S. New J. Chem. 2020, 44, 15321–15336. doi:10.1039/d0nj03450a
Return to citation in text: [1] -
Sinha, A. K.; Mondal, B.; Kundu, M.; Chakraborty, B.; Roy, U. K. Org. Chem. Front. 2014, 1, 1270–1275. doi:10.1039/c4qo00235k
Return to citation in text: [1] -
Sun, L.; Sahloul, K.; Mellah, M. ACS Catal. 2013, 3, 2568–2573. doi:10.1021/cs400587s
Return to citation in text: [1] -
de Souza, R. F. M.; Areias, M. C. C.; Bieber, L. W.; Navarro, M. Green Chem. 2011, 13, 1118–1120. doi:10.1039/c0gc00947d
Return to citation in text: [1] -
Zhang, L.; Zha, Z.; Zhang, Z.; Li, Y.; Wang, Z. Chem. Commun. 2010, 46, 7196–7198. doi:10.1039/c0cc01964j
Return to citation in text: [1] -
Huang, J.-M.; Ren, H.-R. Chem. Commun. 2010, 46, 2286–2288. doi:10.1039/b922897g
Return to citation in text: [1] -
Zhang, L.; Zha, Z.; Wang, Z.; Fu, S. Tetrahedron Lett. 2010, 51, 1426–1429. doi:10.1016/j.tetlet.2010.01.026
Return to citation in text: [1] -
Huang, J.-M.; Dong, Y. Chem. Commun. 2009, 3943–3945. doi:10.1039/b905553c
Return to citation in text: [1] -
Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h
Return to citation in text: [1] [2] [3] [4] [5] -
Durandetti, M.; Meignein, C.; Périchon, J. J. Org. Chem. 2003, 68, 3121–3124. doi:10.1021/jo026782r
Return to citation in text: [1] -
Hilt, G.; Smolko, K. I. Angew. Chem., Int. Ed. 2001, 40, 3399–3402. doi:10.1002/1521-3773(20010917)40:18<3399::aid-anie3399>3.0.co;2-t
Return to citation in text: [1] -
Rollin, Y.; Derien, S.; Duñach, E.; Gebehenne, C.; Perichon, J. Tetrahedron 1993, 49, 7723–7732. doi:10.1016/s0040-4020(01)87246-x
Return to citation in text: [1] -
Hebri, H.; Duñach, E.; Périchon, J. Tetrahedron Lett. 1993, 34, 1475–1478. doi:10.1016/s0040-4039(00)60322-2
Return to citation in text: [1] -
Tokuda, M.; Uchida, M.; Katoh, Y.; Suginome, H. Chem. Lett. 1990, 19, 461–462. doi:10.1246/cl.1990.461
Return to citation in text: [1] -
Tokuda, M.; Satoh, S.; Suginome, H. J. Org. Chem. 1989, 54, 5608–5613. doi:10.1021/jo00284a040
Return to citation in text: [1] -
Durandetti, S.; Sibille, S.; Perichon, J. J. Org. Chem. 1989, 54, 2198–2204. doi:10.1021/jo00270a033
Return to citation in text: [1] -
Minato, M.; Tsuji, J. Chem. Lett. 1988, 17, 2049–2052. doi:10.1246/cl.1988.2049
Return to citation in text: [1] -
Uneyama, K.; Matsuda, H.; Torii, S. Tetrahedron Lett. 1984, 25, 6017–6020. doi:10.1016/s0040-4039(01)81748-2
Return to citation in text: [1] -
Lapeña, D.; Lomba, L.; Artal, M.; Lafuente, C.; Giner, B. Fluid Phase Equilib. 2019, 492, 1–9. doi:10.1016/j.fluid.2019.03.018
Return to citation in text: [1] -
Niharika, P.; Ramulu, B. V.; Satyanarayana, G. ACS Omega 2018, 3, 218–228. doi:10.1021/acsomega.7b01553
Return to citation in text: [1] [2] [3] -
Dam, J. H.; Fristrup, P.; Madsen, R. J. Org. Chem. 2008, 73, 3228–3235. doi:10.1021/jo800180d
Return to citation in text: [1] -
Smith, K.; Lock, S.; El-Hiti, G. A.; Wada, M.; Miyoshi, N. Org. Biomol. Chem. 2004, 2, 935–938. doi:10.1039/b400179f
Return to citation in text: [1]
| 50. | Niharika, P.; Ramulu, B. V.; Satyanarayana, G. ACS Omega 2018, 3, 218–228. doi:10.1021/acsomega.7b01553 |
| 39. | Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h |
| 50. | Niharika, P.; Ramulu, B. V.; Satyanarayana, G. ACS Omega 2018, 3, 218–228. doi:10.1021/acsomega.7b01553 |
| 1. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 51. | Dam, J. H.; Fristrup, P.; Madsen, R. J. Org. Chem. 2008, 73, 3228–3235. doi:10.1021/jo800180d |
| 17. | Smith, E. L.; Abbott, A. P.; Ryder, K. S. Chem. Rev. 2014, 114, 11060–11082. doi:10.1021/cr300162p |
| 18. | Hansen, B. B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J. M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B. W.; Gurkan, B.; Maginn, E. J.; Ragauskas, A.; Dadmun, M.; Zawodzinski, T. A.; Baker, G. A.; Tuckerman, M. E.; Savinell, R. F.; Sangoro, J. R. Chem. Rev. 2021, 121, 1232–1285. doi:10.1021/acs.chemrev.0c00385 |
| 39. | Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h |
| 16. | Bornemann, S.; Handy, S. T. Molecules 2011, 16, 5963–5974. doi:10.3390/molecules16075963 |
| 39. | Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h |
| 2. | Nozaki, S.; Suzuki, Y.; Goto, T. Electrochim. Acta 2024, 493, 144431. doi:10.1016/j.electacta.2024.144431 |
| 3. | Kong, X.; Liu, Q.; Chen, Y.; Wang, W.; Chen, H.-F.; Wang, W.; Zhang, S.; Chen, X.; Cao, Z.-Y. Green Chem. 2024, 26, 3435–3440. doi:10.1039/d3gc04528e |
| 4. | David, M.; Galli, E.; Brown, R. C. D.; Feroci, M.; Vetica, F.; Bortolami, M. Beilstein J. Org. Chem. 2023, 19, 1966–1981. doi:10.3762/bjoc.19.147 |
| 5. | Witherspoon, E.; Ling, P.; Winchester, W.; Zhao, Q.; Ibrahim, A.; Riley, K. E.; Wang, Z. ACS Omega 2022, 7, 42828–42834. doi:10.1021/acsomega.2c04748 |
| 6. | Rocco, D.; Chiarotto, I.; Mattiello, L.; Pandolfi, F.; Zane, D.; Feroci, M. Pure Appl. Chem. 2019, 91, 1709–1715. doi:10.1515/pac-2018-1118 |
| 7. | Yimin, D.; Lanli, N.; Hui, L.; Jiaqi, Z.; Linping, Y.; Qiuju, F. Int. J. Electrochem. Sci. 2018, 13, 1084–1095. doi:10.20964/2018.01.85 |
| 8. | Kathiresan, M.; Velayutham, D. Chem. Commun. 2015, 51, 17499–17516. doi:10.1039/c5cc06961k |
| 9. | Zhao, S.-F.; Horne, M.; Bond, A. M.; Zhang, J. Phys. Chem. Chem. Phys. 2015, 17, 19247–19254. doi:10.1039/c5cp00095e |
| 10. | Kronenwetter, H.; Husek, J.; Etz, B.; Jones, A.; Manchanayakage, R. Green Chem. 2014, 16, 1489–1495. doi:10.1039/c3gc41641k |
| 11. | Orsini, M.; Chiarotto, I.; Feeney, M. M. M.; Feroci, M.; Sotgiu, G.; Inesi, A. Electrochem. Commun. 2011, 13, 738–741. doi:10.1016/j.elecom.2011.04.025 |
| 12. | Liu, Y.-Z.; Lin, M.-Y.; Xiao, L.-P.; Zhang, K.; Lu, J.-X. Chin. J. Chem. 2008, 26, 1168–1172. doi:10.1002/cjoc.200890214 |
| 13. | Mellah, M.; Zeitouny, J.; Gmouh, S.; Vaultier, M.; Jouikov, V. Electrochem. Commun. 2005, 7, 869–874. doi:10.1016/j.elecom.2005.06.002 |
| 14. | Doherty, A. P.; Brooks, C. A. Electrochim. Acta 2004, 49, 3821–3826. doi:10.1016/j.electacta.2003.12.058 |
| 15. | Mellah, M.; Gmouh, S.; Vaultier, M.; Jouikov, V. Electrochem. Commun. 2003, 5, 591–593. doi:10.1016/s1388-2481(03)00136-x |
| 50. | Niharika, P.; Ramulu, B. V.; Satyanarayana, G. ACS Omega 2018, 3, 218–228. doi:10.1021/acsomega.7b01553 |
| 28. | Yusof, R.; Abdulmalek, E.; Sirat, K.; Rahman, M. B. A. Molecules 2014, 19, 8011–8026. doi:10.3390/molecules19068011 |
| 49. | Lapeña, D.; Lomba, L.; Artal, M.; Lafuente, C.; Giner, B. Fluid Phase Equilib. 2019, 492, 1–9. doi:10.1016/j.fluid.2019.03.018 |
| 27. | Gonzalez-Gallardo, N.; Saavedra, B.; Guillena, G.; Ramón, D. J. Appl. Organomet. Chem. 2021, 35, e6418. doi:10.1002/aoc.6418 |
| 25. | Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207–2293. doi:10.1021/cr00022a010 |
| 26. | Denmark, S. E.; Fu, J. Chem. Rev. 2003, 103, 2763–2794. doi:10.1021/cr020050h |
| 39. | Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h |
| 20. | Mousa, M. O.; Adly, M. E.; Mahmoud, A. M.; El-Nassan, H. B. ACS Omega 2024, 9, 14198–14209. doi:10.1021/acsomega.3c09790 |
| 21. | El-Nassan, H. B.; El-Mosallamy, S. S.; Mahmoud, A. M. Sustainable Chem. Pharm. 2023, 35, 101207. doi:10.1016/j.scp.2023.101207 |
| 22. | Osman, E. O.; Mahmoud, A. M.; El-Mosallamy, S. S.; El-Nassan, H. B. J. Electroanal. Chem. 2022, 920, 116629. doi:10.1016/j.jelechem.2022.116629 |
| 23. | Trujillo, S. A.; Peña-Solórzano, D.; Bejarano, O. R.; Ochoa-Puentes, C. RSC Adv. 2020, 10, 40552–40561. doi:10.1039/d0ra06871c |
| 24. | Golgovici, F.; Anicai, L.; Florea, A.; Visan, T. Curr. Nanosci. 2020, 16, 478–494. doi:10.2174/1573413715666190206145036 |
| 29. | Zhang, J.; Zhang, L.; Xie, W.; Chen, M.; Zhang, C.; Qin, Y.; Zhao, J.; Wang, F.; Liu, Z.-Q. Green Chem. 2024, 26, 7002–7006. doi:10.1039/d4gc01838a |
| 30. | Zhang, Q.; Liang, K.; Guo, C. Angew. Chem., Int. Ed. 2022, 61, e202210632. doi:10.1002/anie.202210632 |
| 31. | Torabi, S.; Jamshidi, M.; Amooshahi, P.; Mehrdadian, M.; Khazalpour, S. New J. Chem. 2020, 44, 15321–15336. doi:10.1039/d0nj03450a |
| 32. | Sinha, A. K.; Mondal, B.; Kundu, M.; Chakraborty, B.; Roy, U. K. Org. Chem. Front. 2014, 1, 1270–1275. doi:10.1039/c4qo00235k |
| 33. | Sun, L.; Sahloul, K.; Mellah, M. ACS Catal. 2013, 3, 2568–2573. doi:10.1021/cs400587s |
| 34. | de Souza, R. F. M.; Areias, M. C. C.; Bieber, L. W.; Navarro, M. Green Chem. 2011, 13, 1118–1120. doi:10.1039/c0gc00947d |
| 35. | Zhang, L.; Zha, Z.; Zhang, Z.; Li, Y.; Wang, Z. Chem. Commun. 2010, 46, 7196–7198. doi:10.1039/c0cc01964j |
| 36. | Huang, J.-M.; Ren, H.-R. Chem. Commun. 2010, 46, 2286–2288. doi:10.1039/b922897g |
| 37. | Zhang, L.; Zha, Z.; Wang, Z.; Fu, S. Tetrahedron Lett. 2010, 51, 1426–1429. doi:10.1016/j.tetlet.2010.01.026 |
| 38. | Huang, J.-M.; Dong, Y. Chem. Commun. 2009, 3943–3945. doi:10.1039/b905553c |
| 39. | Zha, Z.; Hui, A.; Zhou, Y.; Miao, Q.; Wang, Z.; Zhang, H. Org. Lett. 2005, 7, 1903–1905. doi:10.1021/ol050483h |
| 40. | Durandetti, M.; Meignein, C.; Périchon, J. J. Org. Chem. 2003, 68, 3121–3124. doi:10.1021/jo026782r |
| 41. | Hilt, G.; Smolko, K. I. Angew. Chem., Int. Ed. 2001, 40, 3399–3402. doi:10.1002/1521-3773(20010917)40:18<3399::aid-anie3399>3.0.co;2-t |
| 42. | Rollin, Y.; Derien, S.; Duñach, E.; Gebehenne, C.; Perichon, J. Tetrahedron 1993, 49, 7723–7732. doi:10.1016/s0040-4020(01)87246-x |
| 43. | Hebri, H.; Duñach, E.; Périchon, J. Tetrahedron Lett. 1993, 34, 1475–1478. doi:10.1016/s0040-4039(00)60322-2 |
| 44. | Tokuda, M.; Uchida, M.; Katoh, Y.; Suginome, H. Chem. Lett. 1990, 19, 461–462. doi:10.1246/cl.1990.461 |
| 45. | Tokuda, M.; Satoh, S.; Suginome, H. J. Org. Chem. 1989, 54, 5608–5613. doi:10.1021/jo00284a040 |
| 46. | Durandetti, S.; Sibille, S.; Perichon, J. J. Org. Chem. 1989, 54, 2198–2204. doi:10.1021/jo00270a033 |
| 47. | Minato, M.; Tsuji, J. Chem. Lett. 1988, 17, 2049–2052. doi:10.1246/cl.1988.2049 |
| 48. | Uneyama, K.; Matsuda, H.; Torii, S. Tetrahedron Lett. 1984, 25, 6017–6020. doi:10.1016/s0040-4039(01)81748-2 |
| 52. | Smith, K.; Lock, S.; El-Hiti, G. A.; Wada, M.; Miyoshi, N. Org. Biomol. Chem. 2004, 2, 935–938. doi:10.1039/b400179f |
© 2024 Taylor and Handy; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.