Abstract
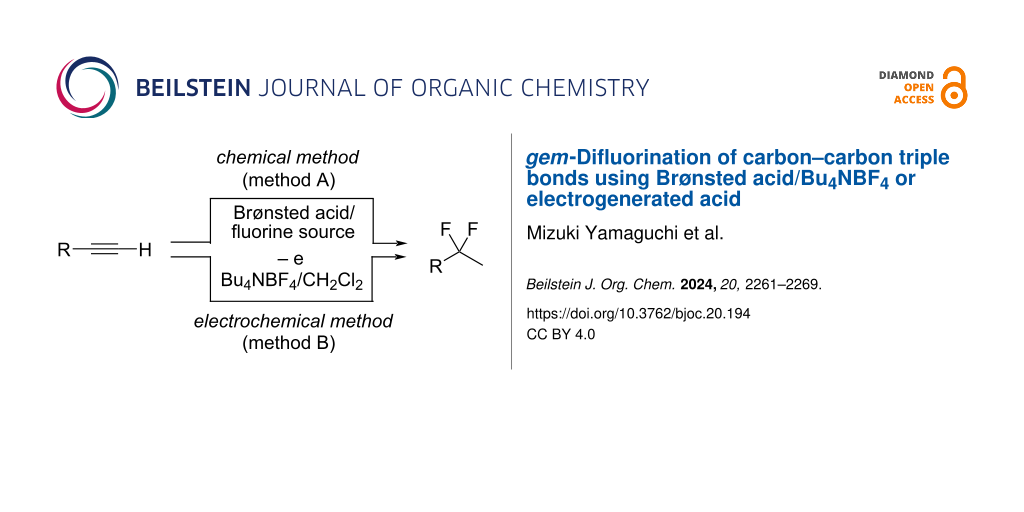
gem-Difluorination of carbon–carbon triple bonds was conducted using Brønsted acids, such as Tf2NH and TfOH, combined with Bu4NBF4 as the fluorine source. The electrochemical oxidation of a Bu4NBF4/CH2Cl2 solution containing alkyne substrates could also give the corresponding gem-difluorinated compounds (in-cell method). The ex-cell electrolysis method was also applicable for gem-difluorination of alkynes.

Graphical Abstract
Introduction
Organofluorine compounds have attracted great attention in various fields, such as organic materials and pharmaceuticals [1-3], because fluorinated compounds sometimes show specific properties [4]. So far, several methods have been developed for the synthesis of fluorinated compounds. Using nucleophilic fluorinating reagents, such as diethylaminosulfur trifluoride (DAST), HF, CsF, and AgF has been established as a reliable method. Electrophilic fluorinating reagents, such as 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor), N-fluorobenzenesulfonimide, and fluorobenziodoxole, are also utilized as F+ equivalents to introduce fluorine atoms into organic molecules. In addition, various trifluoromethylation reagents have been developed so far [5-18]. Transition-metal-catalyzed fluorination and trifluoromethylation methods have also been proposed [19,20]. Thus, the synthesis of fluorinated compounds is an active research field. Among these compounds, skeletons bearing CF2 units are important [21-24], because such molecules can change the physical properties and biological activity. They can also serve as building blocks for further transformations.
We have focused on the investigation of gem-difluorination of carbon–carbon triple bonds, because this procedure is one of the most simple but powerful and straightforward methods. In addition, there have been a few reports in the literature that seem to mainly rely on the use of HF or its complexes as a reagent. These reactions seem to proceed via the formation of the vinyl fluoride as the intermediate [25-28].
In the first example, Olah and co-workers reported the reaction of terminal alkynes with HF/pyridine (Olah reagent) (Figure 1, reaction 1) [29-32], although the original work was developed by Linn and Plueddeman using HF [33-35]. As another example, Renoux and co-workers developed the utility of SbF5/HF (Figure 1, reaction 2) [36]. In 2014, the HF/N,N’-dimethylpropyleneurea (DMPU) complex in the presence of an Au catalyst was found to be a good reagent for the gem-difluorination of alkynes, reported by Hammond and Xu (Figure 1, reaction 3) [37]. HF/DMPU is easy to handle under experimental conditions. In addition, they recently reported the utilization of a combination of KHSO4·13HF and DMPU·12HF under neat conditions for the gem-difluorination of alkynes (Figure 1, reaction 4) [38]. In 2020, the utility of 2,6-dichloropyridinium tetrafluoroborate was nicely demonstrated for the gem-difluorination by Liu and Wang (Figure 1, reaction 5) [39].
Although some procedures have been reported, the use of hazardous reagents such as HF is still inevitable [40,41]. Quite recently, Crimmin and co-workers reported gem-difluorination by shuttling between fluoroalkanes and alkynes, in which catalytic HF played a key role [42]. In the course of our study on the fluorination reaction, we have envisioned that the combination of a Brønsted acid, such as Tf2NH and TfOH, with Bu4NBF4 might be effective to promote the gem-difluorination of alkynes because of the in situ generation of HF equivalents (Figure 1, reaction 6, chemical method). In addition, the electrogenerated acid (EGA) [43-52] from a solution of Bu4NBF4/CH2Cl2 containing substrates might also promote the same reactions (Figure 1, reaction 6, electrochemical method). Currently, electrochemistry can be regarded as a promising technique in organic synthesis, because heavy-metal reagents can be avoided for the oxidation or reduction of organic molecules. Herein, we wish to report that the combination of the excess amount of Brønsted acid and Bu4NBF4 or the use of an EGA in Bu4NBF4/CH2Cl2 can serve as suitable reagents for the gem-difluorination of alkynes. These procedures are practical, simple and environmentally friendly, because HF or its equivalent is indirectly prepared and utilized in only solution phase.
![[1860-5397-20-194-1]](/bjoc/content/figures/1860-5397-20-194-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: gem-Difluorination of carbon–carbon triple bonds. Selected examples from (1) to (5), and this work of (6).
Figure 1: gem-Difluorination of carbon–carbon triple bonds. Selected examples from (1) to (5), and this work ...
Results and Discussion
First, we have chosen hex-5-yn-1-ylbenzene (1a) as the model substrate in the reaction optimization (Table 1, method A). The reaction was carried out as follows: Hex-5-yn-1-ylbenzene (1a, 0.5 mmol) was reacted with the Brønsted acid (X equiv) and the fluorine source (Y equiv) in the solvent (4 mL) at temperature of T (°C) for Z hours. The chemical yield of the desired product, (5,5-difluorohexyl)benzene (2a), was evaluated for reaction optimization by using the 19F nuclear magnetic resonance (NMR) yield, in which trifluorotoluene (CF3C6H5) was used as an internal standard. The use of Tf2NH (5 equiv or 10 equiv) and Bu4NBF4 (5 equiv) in CH2Cl2 at room temperature gave the corresponding product 2a in up to 83% yield (Table 1, entries 1–5). The use of CF3COOH did not yield 2a at all (Table 1, entry 6), but TfOH gave the product 2a in 72% yield (Table 1, entry 7). As for the solvent, CH3CN slightly afforded 2a, although N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were not suitable for the reactions (Table 1, entries 8–10). A fluorine source, such as Bu4NF or BF3·Et2O, instead of Bu4NBF4 was not effective (Table 1, entries 11 and 12). Finally, investigations of the amount of Bu4NBF4 and the reaction temperature demonstrated that conditions including Bu4NBF4 (9 equiv) and room temperature gave the best result (Table 1, entries 13–17). Based on the above investigation, we decided to use the optimized conditions in entry 2, because reducing the amount of Bu4NBF4, for example, to 5 equiv is important from the viewpoint of eco-friendly chemical synthesis. The reaction time of 16 h also seems to be suitable for the investigation. Thus, the combination of Tf2NH and Bu4NBF4 might generate HBF4 in the solution.
Table 1: Optimization of the gem-difluorination of hex-5-yn-1-ylbenzene (1a) to form difluorinated compound 2a (method A).
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-194-i4.svg?max-width=637&scale=1.0)
|
||||||||
| Entry |
Brønsted
acid |
X
(equiv) |
Fluorine
source |
Y
(equiv) |
Solvent |
Reaction
time Z (h) |
T (°C) | % Yielda |
| 1 | Tf2NH | 5 | Bu4NBF4 | 5 | CH2Cl2 | 8 | rt | 83 |
| 2 | Tf2NH | 5 | Bu4NBF4 | 5 | CH2Cl2 | 16 | rt | 83 (72)b |
| 3 | Tf2NH | 5 | Bu4NBF4 | 5 | CH2Cl2 | 24 | rt | 82 |
| 4 | Tf2NH | 3 | Bu4NBF4 | 5 | CH2Cl2 | 16 | rt | 75 |
| 5 | Tf2NH | 10 | Bu4NBF4 | 5 | CH2Cl2 | 16 | rt | 46 |
| 6 | CF3COOH | 5 | Bu4NBF4 | 5 | CH2Cl2 | 16 | rt | n.r.c |
| 7 | TfOH | 5 | Bu4NBF4 | 5 | CH2Cl2 | 16 | rt | 72 |
| 8 | Tf2NH | 5 | Bu4NBF4 | 5 | CH3CN | 16 | rt | 6 |
| 9 | Tf2NH | 5 | Bu4NBF4 | 5 | DMF | 16 | rt | n.r.c |
| 10 | Tf2NH | 5 | Bu4NBF4 | 5 | DMSO | 16 | rt | n.r.c |
| 11d | Tf2NH | 5 | Bu4NF | 5 | CH2Cl2 | 16 | rt | n.r.c |
| 12 | Tf2NH | 5 | BF3·Et2O | 5 | CH2Cl2 | 16 | rt | n.d.e |
| 13 | Tf2NH | 5 | Bu4NBF4 | 3 | CH2Cl2 | 16 | rt | 50 |
| 14 | Tf2NH | 5 | Bu4NBF4 | 9 | CH2Cl2 | 16 | rt | 89 |
| 15 | Tf2NH | 5 | Bu4NBF4 | 9 | CH2Cl2 | 16 | 0 | 31 |
| 16 | Tf2NH | 5 | Bu4NBF4 | 9 | CH2Cl2 | 16 | −40 | n.r.c |
| 17 | Tf2NH | 5 | Bu4NBF4 | 9 | CH2Cl2 | 16 | 40 | 82 |
a19F NMR yields. Trifluorotoluene (CF3C6H5) was used as an internal standard. bIsolated yield after silica gel column chromatography of crude product. cn.r. = no reaction. dA solution of Bu4NF/THF underwent vacuum drying to prepare Bu4NF without THF. Then, CH2Cl2 was added to Bu4NF to prepare a solution. en.d. = not detected.
Next, the electrochemical method was studied for gem-difluorination. In a previous report by us, we found that the electrogenerated acids of “H+BF4−” equivalents can actually serve as H+ equivalents [51,52]. We have envisioned that electrogenerated acids such as ’’H+BF4−’’ equivalents might serve as good reagents for the gem-difluorination of alkynes. Thus, we have examined the electrochemical oxidation of a solution of Bu4NBF4/CH2Cl2 containing 1a (0.5 mmol) in a divided cell using 8 mA or 16 mA (Scheme 1, method B, in-cell method). In-cell method means that EGA was generated in the presence of the substrate. The total electricity of 3.0 F/mol vs 1a was passed to the solution. Interestingly, the result gave the corresponding difluorinated compound 2a in 29% yield in the case of 8 mA, as shown by 19F NMR analysis. In addition, 2a was obtained in 42% yield by 19F NMR analysis in the case of 16 mA [53].
![[1860-5397-20-194-i1]](/bjoc/content/inline/1860-5397-20-194-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: gem-Difluorination promoted by electrogenerated acids (method B).
Scheme 1: gem-Difluorination promoted by electrogenerated acids (method B).
With the successful formation of (5,5-difluorohexyl)benzene (2a) by the chemical (method A) and electrochemical oxidation (method B) methods in hand, we have investigated the scope and limitations of gem-difluorination for various alkynes (Table 2). Electrochemical oxidation of method B was conducted by using 8 mA. The reaction of but-3-yn-1-ylbenzene (1b) in method A gave the corresponding compound 2b in 21% isolated yield (Table 2, entry 1). The 19F NMR result indicated 63% yield. Because of the low molecular weight of 2b, the isolated yield might be somewhat lower. In contrast, method B produced 2b in 6% isolated yield (Table 2, entry 2). The 19F NMR result indicated 29% yield. As for the internal carbon–carbon triple bonds, diphenylacetylene (1c) was tested, but the desired product 2c was not obtained in any of the two methods (Table 2, entries 3 and 4). In the case of an aliphatic terminal alkyne, such as dec-1-yne (1d), the 19F NMR study indicated 46% yield with method A (Table 2, entry 5), but it was difficult to purify and isolate product 2d because of the low molecular weight. Scale up conditions of method A, for the purpose of the isolation, led to the formation of the corresponding product 2d in 40% yield as the 19F NMR analysis (Table 2, entry 6), but the isolation of 2d was difficult [54]. Method B gave 2d in 35% yield, as shown by the 19F NMR analysis (Table 2, entry 7). Another alkyne, namely, octadec-1-yne (1e), was found to be a nice substrate for gem-difluorination to yield the difluorinated compound 2e (Table 2, entries 8 and 9). Interestingly, terminal alkynes bearing –OH and –O– functional groups, such as 1f and 1g, were used for reactions, and the corresponding products 2f and 2g were obtained by both methods (Table 2, entries 10–13). In addition, 2-(pent-4-yn-1-yl)isoindoline-1,3-dione (1h) was utilized for the construction of the CF2 unit under the same conditions to give 2h (Table 2, entries 14 and 15). The substrate bearing a halogen, such as 10-iododec-1-yne (1i) in method A, produced the corresponding 2i in 60% isolated yield (Table 2, entry 16). Likewise, method B also gave 2i in 21% yield, as shown by the 19F NMR analysis (Table 2, entry 17), but it was difficult to purify and isolate the product 2i in this case. Finally, the internal aliphatic alkyne such as dodec-6-yne (1j) was found to be effective for the gem-difluorination. Method A gave 2j in 38% isolated yield, and method B produced 2j in 10% isolated yield (Table 2, entries 18 and 19).
Table 2: Scope and limitations.
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-194-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | Product | Method | % Yielda |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-194-i6.svg?max-width=637&scale=1.0)
1b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-194-i7.svg?max-width=637&scale=1.0)
2b |
A | 21 (63) |
| 2 | B | 6 (29) | ||
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-194-i8.svg?max-width=637&scale=1.0)
1c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-194-i9.svg?max-width=637&scale=1.0)
2c |
A | n.d.b |
| 4 | B | n.d.b | ||
| 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-20-194-i10.svg?max-width=637&scale=1.0)
1d |
![[Graphic 8]](/bjoc/content/inline/1860-5397-20-194-i11.svg?max-width=637&scale=1.0)
2d |
A | –c (46) |
| 6 | A | 4c (40)d | ||
| 7 | B | –c (35) | ||
| 8 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-20-194-i12.svg?max-width=637&scale=1.0)
1e |
![[Graphic 10]](/bjoc/content/inline/1860-5397-20-194-i13.svg?max-width=637&scale=1.0)
2e |
A | 67 (86) |
| 9 | B | 50e (45) | ||
| 10 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-20-194-i14.svg?max-width=637&scale=1.0)
1f |
![[Graphic 12]](/bjoc/content/inline/1860-5397-20-194-i15.svg?max-width=637&scale=1.0)
2f |
A | 47 (59) |
| 11 | B | 41e (37) | ||
| 12 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-20-194-i16.svg?max-width=637&scale=1.0)
1g |
![[Graphic 14]](/bjoc/content/inline/1860-5397-20-194-i17.svg?max-width=637&scale=1.0)
2g |
A | 58e (70) |
| 13 | B | 40e (46) | ||
| 14f |
![[Graphic 15]](/bjoc/content/inline/1860-5397-20-194-i18.svg?max-width=637&scale=1.0)
1h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-20-194-i19.svg?max-width=637&scale=1.0)
2h |
A | 37 (55) |
| 15 | B | 15 (13) | ||
| 16 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-20-194-i20.svg?max-width=637&scale=1.0)
1i |
![[Graphic 18]](/bjoc/content/inline/1860-5397-20-194-i21.svg?max-width=637&scale=1.0)
2i |
A | 60 (81) |
| 17 | B | –c (21) | ||
| 18 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-20-194-i22.svg?max-width=637&scale=1.0)
1j |
![[Graphic 20]](/bjoc/content/inline/1860-5397-20-194-i23.svg?max-width=637&scale=1.0)
2j |
A | 38 (62) |
| 19 | B | 10 (24) | ||
aIsolated yields. Silica gel column chromatography and/or preparative GPC separation were/was conducted for the purification. 19F NMR yields of the crude products are shown in parentheses. bn.d. = not detected. cIt was impossible to purify and isolate the corresponding product, although the product was confirmed by NMR analysis, when the crude product was prepared. The reason might be due to volatility derived from the low molecular weight. dThe reaction was conducted in the fourfold scale. eIsolated products contained a small amount of impurity. fThe conditions such as Bu4NBF4 (9 equiv) and Tf2NH (5 equiv) in CH2Cl2 at 40 °C for 16 h were used.
Another procedure involving electrochemical oxidation was also applied (the ex-cell method) [55,56]. Ex-cell method means that EGA was generated in the absence of the substrate, and the substrate was added to the solution after the electrolysis. Optimized conditions and the result are described in Scheme 2. Namely, the electrochemical oxidation of a 0.3 M Bu4NBF4/CH2Cl2 solution (8 mL) at 0 °C using 32 mA generated and accumulated the EGA as the pool. An electricity of 6.0 F/mol based on 0.5 mmol was passed to the solution. In order to suppress the increase of the solution temperature under the electrolysis, the electrolysis was conducted at 0 oC. Then, the solution containing EGA was allowed to react with 1a (0.5 mmol) at 0 °C for 0.5 h, giving the corresponding product 2a in 61% yield, as demonstrated by the 19F NMR analysis. The result indicated that CH2Cl2 of the solvent might be oxidized and H+ species (or equivalent units) might be generated by the electrolysis in this case. In addition, ex-cell method can avoid the over-oxidation of 2a, although the excess electricity was passed to the solution.
![[1860-5397-20-194-i2]](/bjoc/content/inline/1860-5397-20-194-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Generation and accumulation of EGA followed by the reaction with 1a for 2a.
Scheme 2: Generation and accumulation of EGA followed by the reaction with 1a for 2a.
A plausible reaction mechanism for the current reactions is described in Scheme 3. The reaction of carbon–carbon triple bonds and H+ species, which are derived from the Brønsted acid (in method A) or EGA (in method B), gives the vinylic carbocation intermediate A, which can react with BF4− to give fluorinated alkene B [57-60]. In the next step, B can undergo the second addition of H+, followed by the incorporation of F− into the carbocation intermediate C, forming the difluorinated compound 2a. The carbocation adjacent to the F atom might be stabilized by the unshared electron pair of F. Thus, the chemical and electrochemical methods we developed here seem to be superior to the conventional method, because the chemical method requires a usual Brønsted acid and solid Bu4NBF4, which can avoid the use of dangerous HF solutions. The electrochemical method also needs only electricity and solid Bu4NBF4, which realizes in situ formation of “HBF4” equivalents.
![[1860-5397-20-194-i3]](/bjoc/content/inline/1860-5397-20-194-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have carried out the gem-difluorination of carbon–carbon triple bonds using Tf2NH/Bu4NBF4 or EGA from Bu4NBF4/CH2Cl2. The feature superiority of these methods is that they do not directly require the use of hazardous HF reagents and expensive metal catalysts. The simple combination of a Brønsted acid with Bu4NBF4 as the fluorine source as well as a simple electrolysis in Bu4NBF4/CH2Cl2 represent new routes to synthesize CF2-incorporated organic molecules from alkynes. Further synthetic applications are in progress in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedure, characterization data of compounds and copies of spectra of 1H NMR and 13C NMR. | ||
| Format: PDF | Size: 2.2 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology.
Return to citation in text: [1] -
Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology.
Return to citation in text: [1] -
O’Hagan, D.; Harper, D. B. J. Fluorine Chem. 1999, 100, 127–133. doi:10.1016/s0022-1139(99)00201-8
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology.
Return to citation in text: [1] -
Krafft, M. P.; Riess, J. G. Chem. Rev. 2009, 109, 1714–1792. doi:10.1021/cr800260k
See for a review for understanding of basic properties for fluorine compounds.
Return to citation in text: [1] -
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds.
Return to citation in text: [1] -
Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds.
Return to citation in text: [1] -
Neumann, C. N.; Ritter, T. Acc. Chem. Res. 2017, 50, 2822–2833. doi:10.1021/acs.accounts.7b00413
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds.
Return to citation in text: [1] -
Koike, T.; Akita, M. Org. Biomol. Chem. 2019, 17, 5413–5419. doi:10.1039/c9ob00734b
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds.
Return to citation in text: [1] -
Umemoto, T. Chem. Rev. 1996, 96, 1757–1778. doi:10.1021/cr941149u
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Hafner, A.; Jung, N.; Bräse, S. Synthesis 2014, 46, 1440–1447. doi:10.1055/s-0033-1341223
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Barata‐Vallejo, S.; Lantaño, B.; Postigo, A. Chem. – Eur. J. 2014, 20, 16806–16829. doi:10.1002/chem.201404005
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Umemoto, T. J. Fluorine Chem. 2014, 167, 3–15. doi:10.1016/j.jfluchem.2014.07.029
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Egami, H.; Sodeoka, M. Angew. Chem., Int. Ed. 2014, 53, 8294–8308. doi:10.1002/anie.201309260
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/c1cc12414e
See for a selected review of fluorinating and trifluoromethylation methods.
Return to citation in text: [1] -
Li, X.; Shi, X.; Li, X.; Shi, D. Beilstein J. Org. Chem. 2019, 15, 2213–2270. doi:10.3762/bjoc.15.218
See for a review of transition metal-catalyzed fluorination and trifluoromethylation.
Return to citation in text: [1] -
Ogiwara, Y.; Sakai, N. Angew. Chem., Int. Ed. 2020, 59, 574–594. doi:10.1002/anie.201902805
See for a review of transition metal-catalyzed fluorination and trifluoromethylation.
Return to citation in text: [1] -
Wu, Y.-b.; Wan, L.; Lu, G.-p.; Cai, C. J. Fluorine Chem. 2018, 206, 125–127. doi:10.1016/j.jfluchem.2017.08.017
See for a recent and selected report for the construction of CF2 units.
Return to citation in text: [1] -
Keereewan, S.; Soorukram, D.; Kuhakarn, C.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2018, 295–305. doi:10.1002/ejoc.201701106
See for a recent and selected report for the construction of CF2 units.
Return to citation in text: [1] -
Masusai, C.; Soorukram, D.; Kuhakarn, C.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2018, 160–169. doi:10.1002/ejoc.201701415
See for a recent and selected report for the construction of CF2 units.
Return to citation in text: [1] -
Lv, W.-X.; Li, Q.; Li, J.-L.; Li, Z.; Lin, E.; Tan, D.-H.; Cai, Y.-H.; Fan, W.-X.; Wang, H. Angew. Chem., Int. Ed. 2018, 57, 16544–16548. doi:10.1002/anie.201810204
See for a recent and selected report for the construction of CF2 units.
Return to citation in text: [1] -
Besset, T.; Poisson, T.; Pannecoucke, X. Eur. J. Org. Chem. 2015, 2765–2789. doi:10.1002/ejoc.201403507
See for a review for vinyl fluorides.
Return to citation in text: [1] -
Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Chem. Soc. Rev. 2011, 40, 2867–2908. doi:10.1039/c0cs00201a
See for a review for vinyl fluorides.
Return to citation in text: [1] -
Che, J.; Li, Y.; Zhang, F.; Zheng, R.; Bai, Y.; Zhu, G. Tetrahedron Lett. 2014, 55, 6240–6242. doi:10.1016/j.tetlet.2014.09.072
See for an example of hydrofluorination of alkynes.
Return to citation in text: [1] -
Gauthier, R.; Mamone, M.; Paquin, J.-F. Org. Lett. 2019, 21, 9024–9027. doi:10.1021/acs.orglett.9b03425
See for an example of hydrofluorination of alkynes.
Return to citation in text: [1] -
Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027
Return to citation in text: [1] -
Olah, G. A.; Li, X.-Y.; Wang, Q.; Prakash, G. K. S. Synthesis 1993, 693–699. doi:10.1055/s-1993-25924
Return to citation in text: [1] -
Olah, G. A.; Mathew, T.; Goeppert, A.; Török, B.; Bucsi, I.; Li, X.-Y.; Wang, Q.; Marinez, E. R.; Batamack, P.; Aniszfeld, R.; Prakash, G. K. S. J. Am. Chem. Soc. 2005, 127, 5964–5969. doi:10.1021/ja0424878
Return to citation in text: [1] -
Yamauchi, Y.; Fukuhara, T.; Hara, S.; Senboku, H. Synlett 2008, 438–442. doi:10.1055/s-2008-1032069
Return to citation in text: [1] -
Grosse, A. V.; Linn, C. B. J. Am. Chem. Soc. 1942, 64, 2289–2292. doi:10.1021/ja01262a019
Return to citation in text: [1] -
Henne, A. L.; Plueddeman, E. P. J. Am. Chem. Soc. 1943, 65, 587–589. doi:10.1021/ja01244a026
Return to citation in text: [1] -
Bello, D.; Cormanich, R. A.; O'Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/ch14298
Return to citation in text: [1] -
Cantet, A.-C.; Carreyre, H.; Gesson, J.-P.; Jouannetaud, M.-P.; Renoux, B. J. Org. Chem. 2008, 73, 2875–2878. doi:10.1021/jo702441p
Return to citation in text: [1] -
Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z
Return to citation in text: [1] -
Lu, Z.; Bajwa, B. S.; Liu, S.; Lee, S.; Hammond, G. B.; Xu, B. Green Chem. 2019, 21, 1467–1471. doi:10.1039/c8gc03876g
Return to citation in text: [1] -
Guo, R.; Qi, X.; Xiang, H.; Geaneotes, P.; Wang, R.; Liu, P.; Wang, Y.-M. Angew. Chem., Int. Ed. 2020, 59, 16651–16660. doi:10.1002/anie.202006278
Return to citation in text: [1] -
Wang, Z.-X.; Livingstone, K.; Hümpel, C.; Daniliuc, C. G.; Mück-Lichtenfeld, C.; Gilmour, R. Nat. Chem. 2023, 15, 1515–1522. doi:10.1038/s41557-023-01344-5
See for a related and recent work.
Return to citation in text: [1] -
Gauthier, R.; Paquin, J.-F. Chem. – Eur. J. 2023, 29, e202301896. doi:10.1002/chem.202301896
See for a recent review.
Return to citation in text: [1] -
Farley, S. E. S.; Mulryan, D.; Rekhroukh, F.; Phanopoulos, A.; Crimmin, M. R. Angew. Chem., Int. Ed. 2024, 63, e202317550. doi:10.1002/anie.202317550
Return to citation in text: [1] -
Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Leech, M. C.; Lam, K. Acc. Chem. Res. 2020, 53, 121–134. doi:10.1021/acs.accounts.9b00586
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018, 118, 4702–4730. doi:10.1021/acs.chemrev.7b00475
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Atobe, M.; Shida, N. Curr. Opin. Electrochem. 2024, 44, 101440. doi:10.1016/j.coelec.2024.101440
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Senboku, H. Chem. Rec. 2021, 21, 2354–2374. doi:10.1002/tcr.202100081
See for a recent review of electro-organic chemistry.
Return to citation in text: [1] -
Uneyama, K.; Isimura, A.; Torii, S. Bull. Chem. Soc. Jpn. 1985, 58, 1859–1860. doi:10.1246/bcsj.58.1859
See for a report of EGAs.
Return to citation in text: [1] -
Kawa, K.; Saitoh, T.; Kaji, E.; Nishiyama, S. Org. Lett. 2013, 15, 5484–5487. doi:10.1021/ol4026342
See for a report of EGAs.
Return to citation in text: [1] -
Matsumoto, K.; Shimazaki, H.; Sanada, T.; Shimada, K.; Hagiwara, S.; Suga, S.; Kashimura, S.; Yoshida, J.-i. Chem. Lett. 2013, 42, 843–845. doi:10.1246/cl.130255
See for a report of EGAs.
Return to citation in text: [1] [2] -
Matsumoto, K.; Shimao, H.; Fujiki, Y.; Kawashita, N.; Kashimura, S. Electrochemistry 2020, 88, 262–264. doi:10.5796/electrochemistry.20-00032
See for a report of EGAs.
Return to citation in text: [1] [2] -
LiBF4 as a fluorine source was not suitable. LiBF4 in CH2Cl2 did not dissolve completely. It was also impossible to pass the electricity in the solution of LiBF4/CH2Cl2.
Return to citation in text: [1] -
The extensive and repetitive investigation for the isolation under the fourfold scale gave 2d in 26% isolated yield as the purified compound.
Return to citation in text: [1] -
Steckhan, E. Angew. Chem., Int. Ed. Engl. 1986, 25, 683–701. doi:10.1002/anie.198606831
See for a review of ex-cell electrochemical synthesis.
Return to citation in text: [1] -
It is difficult to accurately quantify and evaluate EGA in the solution phase.
Return to citation in text: [1] -
Cresswell, A. J.; Davies, S. G.; Roberts, P. M.; Thomson, J. E. Chem. Rev. 2015, 115, 566–611. doi:10.1021/cr5001805
See for a review of BF4− as the F− source.
Return to citation in text: [1] -
Pfeifer, L.; Gouverneur, V. Org. Lett. 2018, 20, 1576–1579. doi:10.1021/acs.orglett.8b00321
See for a recent example for the synthesis of vinyl fluoride compounds.
Return to citation in text: [1] -
Yang, M.-H.; Matikonda, S. S.; Altman, R. A. Org. Lett. 2013, 15, 3894–3897. doi:10.1021/ol401637n
See for a recent example for the synthesis of vinyl fluoride compounds.
Return to citation in text: [1] -
Zhao, M.; Ming, L.; Tang, J.; Zhao, X. Org. Lett. 2016, 18, 416–419. doi:10.1021/acs.orglett.5b03448
See for a recent example for the synthesis of vinyl fluoride compounds.
Return to citation in text: [1]
| 55. |
Steckhan, E. Angew. Chem., Int. Ed. Engl. 1986, 25, 683–701. doi:10.1002/anie.198606831
See for a review of ex-cell electrochemical synthesis. |
| 56. | It is difficult to accurately quantify and evaluate EGA in the solution phase. |
| 53. | LiBF4 as a fluorine source was not suitable. LiBF4 in CH2Cl2 did not dissolve completely. It was also impossible to pass the electricity in the solution of LiBF4/CH2Cl2. |
| 54. | The extensive and repetitive investigation for the isolation under the fourfold scale gave 2d in 26% isolated yield as the purified compound. |
| 1. |
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology. |
| 2. |
Hird, M. Chem. Soc. Rev. 2007, 36, 2070–2095. doi:10.1039/b610738a
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology. |
| 3. |
O’Hagan, D.; Harper, D. B. J. Fluorine Chem. 1999, 100, 127–133. doi:10.1016/s0022-1139(99)00201-8
See for a selected review of fluorine chemistry for organic materials, pharmaceuticals, and chemical biology. |
| 21. |
Wu, Y.-b.; Wan, L.; Lu, G.-p.; Cai, C. J. Fluorine Chem. 2018, 206, 125–127. doi:10.1016/j.jfluchem.2017.08.017
See for a recent and selected report for the construction of CF2 units. |
| 22. |
Keereewan, S.; Soorukram, D.; Kuhakarn, C.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2018, 295–305. doi:10.1002/ejoc.201701106
See for a recent and selected report for the construction of CF2 units. |
| 23. |
Masusai, C.; Soorukram, D.; Kuhakarn, C.; Reutrakul, V.; Pohmakotr, M. Eur. J. Org. Chem. 2018, 160–169. doi:10.1002/ejoc.201701415
See for a recent and selected report for the construction of CF2 units. |
| 24. |
Lv, W.-X.; Li, Q.; Li, J.-L.; Li, Z.; Lin, E.; Tan, D.-H.; Cai, Y.-H.; Fan, W.-X.; Wang, H. Angew. Chem., Int. Ed. 2018, 57, 16544–16548. doi:10.1002/anie.201810204
See for a recent and selected report for the construction of CF2 units. |
| 43. |
Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603
See for a recent review of electro-organic chemistry. |
| 44. |
Leech, M. C.; Lam, K. Acc. Chem. Res. 2020, 53, 121–134. doi:10.1021/acs.accounts.9b00586
See for a recent review of electro-organic chemistry. |
| 45. |
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
See for a recent review of electro-organic chemistry. |
| 46. |
Yoshida, J.-i.; Shimizu, A.; Hayashi, R. Chem. Rev. 2018, 118, 4702–4730. doi:10.1021/acs.chemrev.7b00475
See for a recent review of electro-organic chemistry. |
| 47. |
Atobe, M.; Shida, N. Curr. Opin. Electrochem. 2024, 44, 101440. doi:10.1016/j.coelec.2024.101440
See for a recent review of electro-organic chemistry. |
| 48. |
Senboku, H. Chem. Rec. 2021, 21, 2354–2374. doi:10.1002/tcr.202100081
See for a recent review of electro-organic chemistry. |
| 49. |
Uneyama, K.; Isimura, A.; Torii, S. Bull. Chem. Soc. Jpn. 1985, 58, 1859–1860. doi:10.1246/bcsj.58.1859
See for a report of EGAs. |
| 50. |
Kawa, K.; Saitoh, T.; Kaji, E.; Nishiyama, S. Org. Lett. 2013, 15, 5484–5487. doi:10.1021/ol4026342
See for a report of EGAs. |
| 51. |
Matsumoto, K.; Shimazaki, H.; Sanada, T.; Shimada, K.; Hagiwara, S.; Suga, S.; Kashimura, S.; Yoshida, J.-i. Chem. Lett. 2013, 42, 843–845. doi:10.1246/cl.130255
See for a report of EGAs. |
| 52. |
Matsumoto, K.; Shimao, H.; Fujiki, Y.; Kawashita, N.; Kashimura, S. Electrochemistry 2020, 88, 262–264. doi:10.5796/electrochemistry.20-00032
See for a report of EGAs. |
| 19. |
Li, X.; Shi, X.; Li, X.; Shi, D. Beilstein J. Org. Chem. 2019, 15, 2213–2270. doi:10.3762/bjoc.15.218
See for a review of transition metal-catalyzed fluorination and trifluoromethylation. |
| 20. |
Ogiwara, Y.; Sakai, N. Angew. Chem., Int. Ed. 2020, 59, 574–594. doi:10.1002/anie.201902805
See for a review of transition metal-catalyzed fluorination and trifluoromethylation. |
| 51. |
Matsumoto, K.; Shimazaki, H.; Sanada, T.; Shimada, K.; Hagiwara, S.; Suga, S.; Kashimura, S.; Yoshida, J.-i. Chem. Lett. 2013, 42, 843–845. doi:10.1246/cl.130255
See for a report of EGAs. |
| 52. |
Matsumoto, K.; Shimao, H.; Fujiki, Y.; Kawashita, N.; Kashimura, S. Electrochemistry 2020, 88, 262–264. doi:10.5796/electrochemistry.20-00032
See for a report of EGAs. |
| 5. |
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds. |
| 6. |
Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731–764. doi:10.1021/cr500193b
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds. |
| 7. |
Neumann, C. N.; Ritter, T. Acc. Chem. Res. 2017, 50, 2822–2833. doi:10.1021/acs.accounts.7b00413
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds. |
| 8. |
Koike, T.; Akita, M. Org. Biomol. Chem. 2019, 17, 5413–5419. doi:10.1039/c9ob00734b
See for a selected review for the synthesis of fluorine molecules including CF3-incorporated compounds. |
| 9. |
Umemoto, T. Chem. Rev. 1996, 96, 1757–1778. doi:10.1021/cr941149u
See for a selected review of fluorinating and trifluoromethylation methods. |
| 10. |
Hafner, A.; Jung, N.; Bräse, S. Synthesis 2014, 46, 1440–1447. doi:10.1055/s-0033-1341223
See for a selected review of fluorinating and trifluoromethylation methods. |
| 11. |
Barata‐Vallejo, S.; Lantaño, B.; Postigo, A. Chem. – Eur. J. 2014, 20, 16806–16829. doi:10.1002/chem.201404005
See for a selected review of fluorinating and trifluoromethylation methods. |
| 12. |
Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826–870. doi:10.1021/cr500277b
See for a selected review of fluorinating and trifluoromethylation methods. |
| 13. |
Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h
See for a selected review of fluorinating and trifluoromethylation methods. |
| 14. |
Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386
See for a selected review of fluorinating and trifluoromethylation methods. |
| 15. |
Umemoto, T. J. Fluorine Chem. 2014, 167, 3–15. doi:10.1016/j.jfluchem.2014.07.029
See for a selected review of fluorinating and trifluoromethylation methods. |
| 16. |
Egami, H.; Sodeoka, M. Angew. Chem., Int. Ed. 2014, 53, 8294–8308. doi:10.1002/anie.201309260
See for a selected review of fluorinating and trifluoromethylation methods. |
| 17. |
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
See for a selected review of fluorinating and trifluoromethylation methods. |
| 18. |
Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/c1cc12414e
See for a selected review of fluorinating and trifluoromethylation methods. |
| 40. |
Wang, Z.-X.; Livingstone, K.; Hümpel, C.; Daniliuc, C. G.; Mück-Lichtenfeld, C.; Gilmour, R. Nat. Chem. 2023, 15, 1515–1522. doi:10.1038/s41557-023-01344-5
See for a related and recent work. |
| 41. |
Gauthier, R.; Paquin, J.-F. Chem. – Eur. J. 2023, 29, e202301896. doi:10.1002/chem.202301896
See for a recent review. |
| 4. |
Krafft, M. P.; Riess, J. G. Chem. Rev. 2009, 109, 1714–1792. doi:10.1021/cr800260k
See for a review for understanding of basic properties for fluorine compounds. |
| 42. | Farley, S. E. S.; Mulryan, D.; Rekhroukh, F.; Phanopoulos, A.; Crimmin, M. R. Angew. Chem., Int. Ed. 2024, 63, e202317550. doi:10.1002/anie.202317550 |
| 36. | Cantet, A.-C.; Carreyre, H.; Gesson, J.-P.; Jouannetaud, M.-P.; Renoux, B. J. Org. Chem. 2008, 73, 2875–2878. doi:10.1021/jo702441p |
| 38. | Lu, Z.; Bajwa, B. S.; Liu, S.; Lee, S.; Hammond, G. B.; Xu, B. Green Chem. 2019, 21, 1467–1471. doi:10.1039/c8gc03876g |
| 33. | Grosse, A. V.; Linn, C. B. J. Am. Chem. Soc. 1942, 64, 2289–2292. doi:10.1021/ja01262a019 |
| 34. | Henne, A. L.; Plueddeman, E. P. J. Am. Chem. Soc. 1943, 65, 587–589. doi:10.1021/ja01244a026 |
| 35. | Bello, D.; Cormanich, R. A.; O'Hagan, D. Aust. J. Chem. 2015, 68, 72–79. doi:10.1071/ch14298 |
| 39. | Guo, R.; Qi, X.; Xiang, H.; Geaneotes, P.; Wang, R.; Liu, P.; Wang, Y.-M. Angew. Chem., Int. Ed. 2020, 59, 16651–16660. doi:10.1002/anie.202006278 |
| 29. | Olah, G. A.; Welch, J. T.; Vankar, Y. D.; Nojima, M.; Kerekes, I.; Olah, J. A. J. Org. Chem. 1979, 44, 3872–3881. doi:10.1021/jo01336a027 |
| 30. | Olah, G. A.; Li, X.-Y.; Wang, Q.; Prakash, G. K. S. Synthesis 1993, 693–699. doi:10.1055/s-1993-25924 |
| 31. | Olah, G. A.; Mathew, T.; Goeppert, A.; Török, B.; Bucsi, I.; Li, X.-Y.; Wang, Q.; Marinez, E. R.; Batamack, P.; Aniszfeld, R.; Prakash, G. K. S. J. Am. Chem. Soc. 2005, 127, 5964–5969. doi:10.1021/ja0424878 |
| 32. | Yamauchi, Y.; Fukuhara, T.; Hara, S.; Senboku, H. Synlett 2008, 438–442. doi:10.1055/s-2008-1032069 |
| 57. |
Cresswell, A. J.; Davies, S. G.; Roberts, P. M.; Thomson, J. E. Chem. Rev. 2015, 115, 566–611. doi:10.1021/cr5001805
See for a review of BF4− as the F− source. |
| 58. |
Pfeifer, L.; Gouverneur, V. Org. Lett. 2018, 20, 1576–1579. doi:10.1021/acs.orglett.8b00321
See for a recent example for the synthesis of vinyl fluoride compounds. |
| 59. |
Yang, M.-H.; Matikonda, S. S.; Altman, R. A. Org. Lett. 2013, 15, 3894–3897. doi:10.1021/ol401637n
See for a recent example for the synthesis of vinyl fluoride compounds. |
| 60. |
Zhao, M.; Ming, L.; Tang, J.; Zhao, X. Org. Lett. 2016, 18, 416–419. doi:10.1021/acs.orglett.5b03448
See for a recent example for the synthesis of vinyl fluoride compounds. |
| 25. |
Besset, T.; Poisson, T.; Pannecoucke, X. Eur. J. Org. Chem. 2015, 2765–2789. doi:10.1002/ejoc.201403507
See for a review for vinyl fluorides. |
| 26. |
Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Chem. Soc. Rev. 2011, 40, 2867–2908. doi:10.1039/c0cs00201a
See for a review for vinyl fluorides. |
| 27. |
Che, J.; Li, Y.; Zhang, F.; Zheng, R.; Bai, Y.; Zhu, G. Tetrahedron Lett. 2014, 55, 6240–6242. doi:10.1016/j.tetlet.2014.09.072
See for an example of hydrofluorination of alkynes. |
| 28. |
Gauthier, R.; Mamone, M.; Paquin, J.-F. Org. Lett. 2019, 21, 9024–9027. doi:10.1021/acs.orglett.9b03425
See for an example of hydrofluorination of alkynes. |
| 37. | Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z |
© 2024 Yamaguchi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.