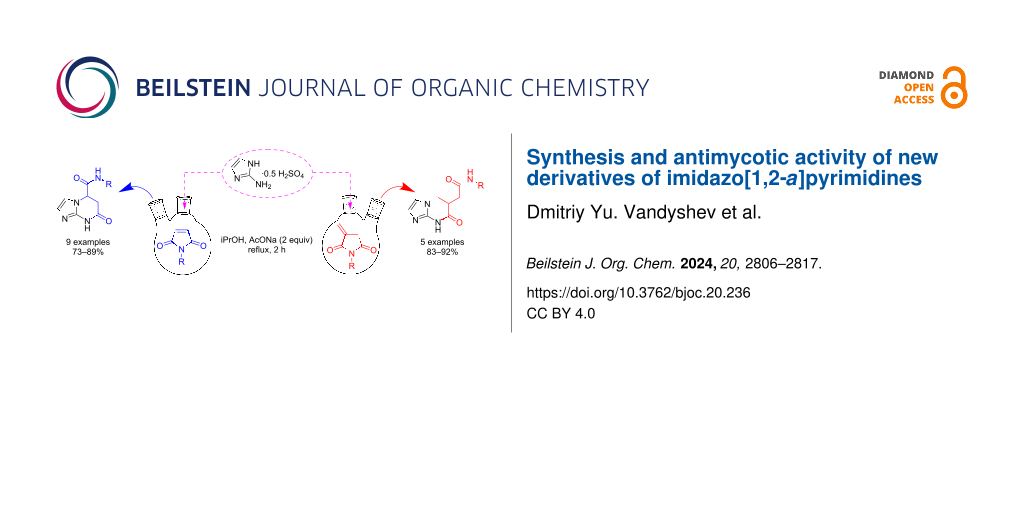
Abstract
The heterocyclic core of imidazo[1,2-a]pyrimidine was formed in satisfactory yields as a result of the interaction of the readily available 2-aminoimidazole with N-substituted maleimides or N-arylitaconimides. The mechanism of the studied processes was postulated basing on experimental data, HPLC–MS analysis of reaction mixtures, and quantum chemical calculations. Molecular docking results of the obtained imidazo[1,2-a]pyrimidines, when compared with voriconazole, a drug already in clinical use, suggest that they may possess antifungal activity against Candida albicans.

Graphical Abstract
Introduction
Nitrogen-containing heterocyclic compounds occupy a pivotal position in the arsenal of modern organic and medicinal chemistry due to their extensive spectrum of physiological activity [1]. Imidazo[1,2-a]pyrimidines represent one of the most promising classes of compounds within this group [2,3]. These small azoheterocyclic frameworks (small-molecule concept) offer a distinctive advantage in the synthesis of new biologically active molecules, as they are synthetic bioisosters of purine bases. Imidazo[1,2-a]pyrimidine derivatives exhibit a wide range of pharmacological properties [4-6]. For example, this scaffold is a key structural element of divaplon [7], fasiplon and taniplon [8], which are anxiolytics and anticonvulsant drugs. However, their application in clinical practice was recently discontinued [9,10]. The use of imidazo[1,2-a]pyrimidine derivatives as effective antifungal agents is worthy of particular attention, as evidenced by the following references [11-15] (Figure 1).
![[1860-5397-20-236-1]](/bjoc/content/figures/1860-5397-20-236-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some biologically active compounds and organic fluorophores containing the imidazo[1,2-a]pyrimidine nucleus.
Figure 1: Some biologically active compounds and organic fluorophores containing the imidazo[1,2-a]pyrimidine...
The best known method for the synthesis of imidazo[1,2-a]pyrimidines is the one developed by Chichibabin [16-19], based on the reaction between 2-aminopyrimidine and α-haloketones. Despite the widespread use of imidazo[1,2-a]pyrimidines obtained by various modifications of this method and a number of others described in detail in the review by Goel et al. [4], literature data on the synthesis of tetrahydroimidazo[1,2-a]pyrimidines without substituents in the second and third positions remain limited. In this context, it is worth mentioning the work of Li and co-workers (2011) [20], who described a single example of the formation of such structures by carrying out an organocatalytic domino aza-Michael–Mannich reaction between benzylidene-1H-imidazol-2-amine and cinnamaldehyde. Although the imidazo[1,2-a]pyrimidines thus formed did not show significant bioactivity, they found applications as additives in electrochemical copper plating processes [21,22] (Figure 2).
![[1860-5397-20-236-2]](/bjoc/content/figures/1860-5397-20-236-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Existing approaches to imidazo[1,2-a]pyrimidines.
Figure 2: Existing approaches to imidazo[1,2-a]pyrimidines.
Based on the above, the main goal of this work was to develop a convenient method for the construction of potentially pharmacophoric imidazo[1,2-a]pyrimidines using readily available derivatives of azaheterocycles as starting materials. As the last ones, we chose the reaction between 2-aminoimidazole and N-arylitaconimides or N-substituted maleimides, which can be analyzed by both classical synthetic and quantum DFT methods. The second problem solved in this work was a preliminary evaluation of the potential bioactivity of the obtained compounds. In particular, a molecular docking experiment to investigate the binding mechanisms to the CYP51 enzyme and an evaluation of the antifungal activity of imidazo[1,2-a]pyrimidines against Candida albicans were performed.
Results and Discussion
N-Arylitaconimides [23] and N-substituted maleimides [24] were used as initial reagents in the synthesis of the target tetrahydroimidazo[1,2-a]pyrimidine derivatives. These compounds are promising sources of C3-synthons and offer the possibility of constructing polysubstituted hydrogenated heterocyclic structures on their basis [25-28], including an acetanilide fragment. The introduction of this fragment into a molecule, often drugs, enhances the cytotoxic, antibacterial, and antiviral activity of the compounds, thus widening the range of their potential applications [28,29] including as antifungal agents [30-33]. These findings are supported by a number of literature sources that highlight the importance of incorporating the acetanilide moiety into potential drugs, including for enhancing the pharmacological activity of imidazo[1,2-a]pyrimidines [1].
It has already been noted that the preferred medium for recyclizations involving imides of itaconic and malic acids by various N,N- and C,N-dinucleophiles, in particular by 1,2-diaminoimidazole derivatives, 5-aminopyrazoles, and 3-aminocyclohexen-2-ones, are mixtures of polar solvents with acetic acid or pure acetic acid [25-28,34-36].
Within the framework of this study, the primary task was to establish the optimal reaction conditions for the interaction of 2-aminoimidazole (1) with N-phenylmaleimide (2a) (Scheme 1). The choice of the investigated reaction conditions was based on the analysis of existing data [28], taking into account experimentally obtained data on the solubility of the starting reagents. The tested solvents were dioxane, PhMe, MeOH, EtOH, iPrOH, MeCN, and DMF (dimethylformamide). Sodium acetate as an additive was necessary for converting 2-aminoimidazole hemisulfate to its basic form in situ. Thin-layer chromatography (TLC) and high-performance liquid chromatography with mass-spectrometric detection (HPLC–MS) were used to monitor the reaction and to identify the products obtained. It should be noted that in the case of HPLC–MS analysis, the signals registered in the mass spectra were interpreted on the basis of pre-calculated weights (as molecular ions with [M + H]+) for all possible initial, intermediate, and expected interaction products (Table 1).
![[1860-5397-20-236-i1]](/bjoc/content/inline/1860-5397-20-236-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of 2-aminoimidazole (1) with N-substituted maleimides (2) and N-arylitaconimides (3).
Scheme 1: Reaction of 2-aminoimidazole (1) with N-substituted maleimides (2) and N-arylitaconimides (3).
Table 1: Screening of reaction conditions for the preparation of 7-oxo-N-phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidine-5-carboxamide (4a).
| Entry | Solvent and additives | Time of the reaction, h | ||||
| 0.5 | 1 | 2 | 3 | 12 | ||
| Amount (%) of 4a in the reaction mixture | ||||||
| 1 | PhMe, AcONa (1 equiv) | 2 | 12 | 18 | 22 | 25 |
| 2 | dioxane, AcONa (1 equiv) | 5 | 15 | 24 | 30 | 35 |
| 3 | DMF, AcONa (1 equiv) | 10 | 10 | 10 | 10 | 10 |
| 4 | MeCN, AcONa (1 equiv) | 12 | 15 | 19 | 19 | 19 |
| 5 | iPrOH, AcONa (1 equiv) | 20 | 45 | 58 | 65 | 65 |
| 6 | MeOH, AcONa (1 equiv) | 8 | 27 | 42 | 48 | 48 |
| 7 | EtOH, AcONa (1 equiv) | 12 | 31 | 46 | 58 | 58 |
| 8 | iPrOH, AcONa (1.5 equiv) | 39 | 57 | 72 | 72 | 72 |
| 9 | iPrOH, AcONa (2 equiv) | 60 | 89 | 89 | 89 | 89 |
| 10 | iPrOH, AcONa (2.5 equiv) | 57 | 85 | 85 | 85 | 85 |
The tentative experiments showed that when toluene or dioxane were used as solvents, the maximum conversion of the reagents was not achieved until after 12 h of boiling, and the yield of the product varied in the range of 25–35% (entries 1 and 2, Table 1). In the cases of DMF and MeCN, the formation of complex, inseparable mixtures of numerous intermediates and products of their subsequent intramolecular cyclization were observed (Table 1, entries 3 and 4).
The best results were obtained when isopropyl alcohol was used, which furnished yields of the final product 4а up to 89% within 1 h (entry 9 in Table 1). In the case of methanol and ethanol, the maximum conversion was also observed during the three-hour boiling, but the yields of the product 4a were significantly lower. It is important to note that the amount of sodium acetate introduced had a significant effect on the rate and completeness of the conversion of the starting materials to the final product (see as an example, entries 8 and 9 in Table 1).
The optimal conditions found for product 4a also proved to be efficient for the preparation of its analogs 5 from N-arylitaconimides 3 (Scheme 1). Therefore, a broad range of variously substituted imidazo[1,2-a]pyrimidines was synthesized (Table 2).
Table 2: Yields of the products 4 and 5.
| Entry | Initial imide | R | Product | Yield, % |
| 1 | 2a | Ph | 4a | 89 |
| 2 | 2b | 4-iPrC6H4 | 4b | 84 |
| 3 | 2c | 2-Me,3-ClC6H3 | 4c | 86 |
| 4 | 2d | 2,5-Cl2C6H3 | 4d | 83 |
| 5 | 2e | 2-Me,5-NO2C6H3 | 4e | 82 |
| 6 | 2f | CH2C6H5 | 4f | 84 |
| 7 | 2g | CH2CH2C6H5 | 4g | 75 |
| 8 | 2h | CH2CH2(4-MeОC6H4) | 4h | 89 |
| 9 | 2i | Me | 4i | 70 |
| 10 | 3a | Ph | 5a | 92 |
| 11 | 3b | 4-BrC6H4 | 5b | 86 |
| 12 | 3c | 4-FC6H4 | 5c | 83 |
| 13 | 3d | 4-ClC6H4 | 5d | 87 |
| 14 | 3e | 4-EtC6H4 | 5e | 88 |
For the interaction of the polyfunctional precursors 1, 2, and 3, two pathways are theoretically possible (Scheme 2 and Scheme 3). The first one involves the N-nucleophilic Michael addition to the activated multiple bond of the imide, leading to the formation of linear intermediates 6 or 8 (pathways A1 and B1) at the expense of the endo-nitrogen atom. The second route involves the participation of an amino group which allows the formation of adducts 7 and 9 (routes A2 and B2).
![[1860-5397-20-236-i2]](/bjoc/content/inline/1860-5397-20-236-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible synthetic routes for the interaction of N-substituted maleimides 2 with 2-aminoimidazole (1).
Scheme 2: Plausible synthetic routes for the interaction of N-substituted maleimides 2 with 2-aminoimidazole (...
![[1860-5397-20-236-i3]](/bjoc/content/inline/1860-5397-20-236-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible synthetic routes for the interaction of or N-arylitaconimides 3 with 2-aminoimidazole (1).
Scheme 3: Plausible synthetic routes for the interaction of or N-arylitaconimides 3 with 2-aminoimidazole (1)....
The intermediates formed then undergo subsequent tandem recyclization of succinimide/citraconimide fragments at the expense of one of the carbonyl groups and the imidazole nucleophilic center not involved in the first step. This process leads to the formation of alternative final products: imidazo[1,2-a]imidazoles 10 and 12, imidazo[1,5-a]pyrimidines 4, 5, 11 and 14, and imidazo[1,2-a]diazines 13 and 15.
The analysis of the spectral data (1H and 13C NMR, 2D NMR spectroscopy, HPLC–HRESIMS) of the products of the investigated reactions unambiguously confirms the processes proceeding along routes A1 and B1 with the formation of N-aryl(alkyl)-7-oxo-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidin-5-carboxamides 4a–i and N-aryl-2-(7-oxo-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidin-6-yl)acetamides 5a–e.
In 1H NMR spectra of the products, the characteristic signals for the protons of the methylene and methine fragments of the pyrimidine cycle are important in establishing the regiochemistry of the process. Thus, in the 1Н NMR spectra of compounds 4, the following reference signals of protons are present: HА-6 (d at δ ≈ 2.70–2.80 ppm, 2J6А,6В = 16.6 Hz and 3J5,6А = 2.8 Hz), HВ-6 (dd at δ ≈ 3.20–3.30 ppm, 2J6А,6В = 16.6 and 3J5,6В = 7.5 Hz), and H-5 (δ = 5.05–5.25 ppm, 3J5,6В = 7.5 Hz 3J5,6А = 2.8 Hz) [37]. It is worth noting that in the spectra of the supposed compounds 11, the HB-5 proton would undergo additional cleavage at the proton of the adjacent NH group of the pyrimidine ring. Additionally, in the probable structures 10 and 12 the protons HA-5 and HA-6 should have been magnetically equivalent. The same reasoning can be applied to the spectra of 5a−e.
The unambiguous assignment of the signals for the methine and methylene groups of compounds 4 and 5 was carried out based on the correlations found in the NOESY 1H,1H and HMBC 1H,13C spectra. As an example, the key correlation interactions for compounds 4d and 5d are depicted in Figure 3.
![[1860-5397-20-236-3]](/bjoc/content/figures/1860-5397-20-236-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Key correlations observed in the NOESY and HMBC spectra of the products 4d and 5d.
Figure 3: Key correlations observed in the NOESY and HMBC spectra of the products 4d and 5d.
Thus, in the NOESY spectra of the imidazopyrimidine 4d there are cross peaks of the methine proton at C-5 with the amide proton of the acetamide fragment (Figure 3), which are not possible in the case of the hypothetical imidazoimidazoles 10 and 12. Also the observed cross peak of the same methine proton with the proton at C-3 of the imidazole core, would not be possible for the alternative imidazopyrimidine system. These results allowed us to reject the structure 11. The lack of correlation in the HMBC spectra between the protons of the amide fragment and the C-5 carbon atom, as well as the presence of cross-peaks of the H-5 proton with the C-8a nodal atom and the C-3 imidazole carbon atom, further supports the formation of product 4d.
Strong interactions between protons of two methylene groups are the most noticeable features in the NOESY spectrum of the imidazo[1,2-a]pyrimidine 5d. Due to the conformational rigidity of its heterocyclic core, only geminal interactions are observed. Hence, the cross peaks can be observed only between two protons of the exo-5-methylene group and between two protons of the endo-methylene group (see Figure 3). Vicinal proton interactions are weakly expressed in the NOESY spectra and the absence of cross peaks between the methine proton and the endocyclic methylene group is apparently due to a conformational effect. Significantly, there are no cross peaks for the protons of the endo-methylene group with the NH proton of the pyrimidine cycle, which would be observed in the alternative structures.
In the HMBC spectrum of the compound 5d, the cross peaks of the endo-methylene protons (CH2-5) with the carbons C-3 and C-8a are the most informative, indicating a close proximity of the interacting nucleus that cannot be realized in the alternative structures (for example, 14 and 15 in Scheme 3). In addition, clearly distinguishable cross-peaks between the protons of the CH2-5 group and both endo and exo-carbonyl groups are observed. This allowed us to explicitly exclude from the analysis structures 13 and 15, which contain both endo-methylene groups, as part of a conformationally rigid seven-membered cycle. In the latter case, the picture would be different: the interaction of both methylene groups with the carbonyl carbon atom would be observed to an equal extent.
In order to obtain additional information on the course of the reaction between 2-aminoimidazole 1 and imides 2 and 3, the minimum energy paths (MEPs) of these processes were calculated. Quantum chemical DFT calculations were performed using the B3LYP/6-311++G(d,p) basis set and taking into account solvation effects using the polarizable continuum model (PCM). Interactions of 1 with N-phenylmaleimide (2a) and N-phenylithaconimide (3a) were considered as model systems (Scheme 4 and Scheme 5).
Based on the calculations performed, the existence of thermodynamically favourable interaction pathways between aminoimidazole 1 and N-phenylmaleimide 2a (pathway A1 and A2) was confirmed, in which intermediates 6a and 7a are formed (Scheme 4). Although intermediate 7a has a lower activation energy (∆G = −0.23 kcal/mol), further recyclization processes are not possible due to the positive free energy change (∆G > 0). In this context, the formation of the final product is only possible to proceed via intermediate 6a, which undergoes subsequent cyclization steps more favorably, leading to the formation of the target product 4a (∆G = −3.02 kcal/mol). This suggests that the first step of intermediate formation is the critical one.
![[1860-5397-20-236-i4]](/bjoc/content/inline/1860-5397-20-236-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Results of MEP calculations for the reaction of N-phenylmaleimide (2a) with 2-aminoimidazole (1).
Scheme 4: Results of MEP calculations for the reaction of N-phenylmaleimide (2a) with 2-aminoimidazole (1).
It is also noteworthy that the Michael addition via intermediate 6a is an irreversible process, given that it is thermodynamically challenging to revert the formed target product 4a to the original starting substances or the corresponding intermediate 6a (∆G(4a→6a) = + 4.04 kcal/mol). Meanwhile, a minimal difference in the free energy of intermediates 6a and 7a may facilitate their interconversion into each other.
A peculiarity of the reaction with N-phenylithaconimide (3a) is that the formation of intermediates 8a and 9a is an endothermic process (paths B1 and B2, correspondingly, Scheme 5). However, the formation of the final product from the starting reagents (∆G = −1.88 kcal/mol) as well as from intermediate 8a (∆G = +3.97 kcal/mol) is thermodynamically more favorable than for all other routes (path B).
![[1860-5397-20-236-i5]](/bjoc/content/inline/1860-5397-20-236-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Results of MEP calculations for the reaction of N-phenylithaconimide (3a) with 2-aminoimidazole (1).
Scheme 5: Results of MEP calculations for the reaction of N-phenylithaconimide (3a) with 2-aminoimidazole (1)....
Fungal infections are a growing threat to public health, resulting in significant economic costs to healthcare systems [38,39]. One of the most common fungal diseases is invasive candidiasis caused by species of the genus Candida [40,41], including C. albicans, C. glabrata, C. tropicalis, and C. parapsilosis. Bloodstream infections caused by these pathogens are associated with high morbidity and mortality, particularly in intensive care patients. Risk factors for invasive candidiasis include the use of immunosuppressive and cytotoxic drugs, broad-spectrum antibiotic therapy, pre-existing illnesses such as AIDS or diabetes, and the use of central venous catheters, urethral catheters, and implantable medical devices [42].
The mainstay of therapy for these infections is the use of broad-spectrum antifungal agents. Among the many drugs and hits, the azole-based systems (including triazoles and imidazoles) are of particular importance in this context. Their mechanism of action is the inhibition of the activity of the enzyme lanosterol 14α-demethylase (CYP51), which is encoded by the CYP51/ERG11 gene. This enzyme contains a haem-like prosthetic group in its active center and is a member of the cytochrome P450 family, which plays a key role in the biosynthesis of sterols. Sterols in turn are integral components of the fungal cell membrane, making inhibition of CYP51 [43] an effective method of controlling fungal infections. The mechanism of inhibition of azoles and their derivatives is based on the formation of a coordination bond between their heterocyclic nitrogen atom, which carries an unshared electron pair, and the haem iron atom. The formation of this bond leads to inhibition of the catalytic activity of CYP51.
A number of previous studies confirmed the efficacy of azole compounds and their derivatives against a variety of strains and clinical isolates of the genus Candida. Nevertheless, the increased use of azole antifungals for the therapy of both mucosal and systemic Candida infections has favored the selection and/or emergence of Candida strains with advanced resistance in patients suffering from life-threatening disseminated forms of the disease, especially in the presence of serious comorbidities or immunodeficiency. The emergence of resistance to azoles [44-46] may prove to be a significant clinical problem, highlighting the importance of finding alternative therapies for fungal infections. Although there are publications on the use of imidazo[1,2-a]pyrimidine derivatives as antifungal agents [47-49], these compounds have not yet demonstrated the desired pharmacological properties. Therefore, the design and synthesis of new compounds of this class with improved ADMET (absorption, distribution, metabolism, excretion, toxicity) pharmacological properties is an urgent task.
Molecular docking was carried out to identify potential binding positions of compounds 4a–e and 5a–e to CYP51 and to evaluate which of these molecules could act as inhibitors of the enzyme. As mentioned above, CYP51 inhibitors contain a heterocyclic nitrogen atom that forms a coordination bond with haem iron. Therefore, only those compounds that could form such a bond according to the molecular docking results were selected as potentially active. Such compounds were the (S)-isomers of compounds 4a–e and the (R)-isomer of compound 5e (Figure 4). No suitable docking solutions were found for the remaining compounds 5a–d in both isomers, the (R)-isomers of compounds 4a–e and the (S)-enantiomer of 5e and therefore, these compounds were classified as inactive.
![[1860-5397-20-236-4]](/bjoc/content/figures/1860-5397-20-236-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Structures of imidazo[1,2-a]pyrimidines selected for docking and voriconazole selected for comparison.
Figure 4: Structures of imidazo[1,2-a]pyrimidines selected for docking and voriconazole selected for comparis...
The docking results are shown in Table 3. Compounds 4a–e bind to the active site of CYP51 with affinities ranging from −7.7 to −8.8 kcal/mol and compound 5e has a much higher affinity of −5.4 kcal/mol. Therefore, we assume that the inhibitor conformation found by docking is energetically unfavorable and therefore the expected activity of compound 5e will be much lower than that of compounds 4a–e.
Table 3: Characterization of ligand–protein interactions for voriconazole and compounds selected during docking.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-236-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-236-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 3]](/bjoc/content/inline/1860-5397-20-236-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-20-236-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-20-236-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-20-236-i11.svg?max-width=637&scale=1.0)
|
Ligand–protein interactions | |||||||||||
| Y122 | F130 | V135 | Y136 | A307 | I373 | L508 | HEM | H374 | I377 | F504 | F234 | ||||||
| Hb | H | H | H | H | H | H | H/MAb | H | H | H | H | ||||||
| V | − | − | 1 | 1 | 2.1 | + | + | + | + | + | + | + | +/+ | − | − | − | − |
| 4b-S | 2 | −8.3 | 0.636 | 0.426 | 2.9 | + | + | − | + | + | + | + | +/+ | + | + | + | − |
| 4c-S | 2 | −7.9 | 0.6 | 0.493 | 3.4 | + | + | − | − | + | + | + | +/+ | + | − | + | − |
| 4a-S | 3 | −8 | 0.667 | 0.424 | 3.5 | + | + | − | − | + | + | + | +/+ | − | − | + | − |
| 4d-S | 2 | −7.7 | 0.667 | 0.478 | 3.5 | + | + | − | − | + | + | + | +/+ | − | − | + | − |
| 4e-S | 1 | −8.8 | 0.667 | 0.416 | 2.8 | + | + | − | − | + | + | + | +/+ | − | − | + | − |
| 5e-R | 18 | −5.4 | 0.417 | 0.478 | 2.5 | + | − | − | − | + | + | + | +/+ | + | + | + | + |
aTanimoto IFP: Tanimoto coefficient for ligand–protein interaction fingerprints. Tanimoto MACCS: Tanimoto coefficient for chemical structure similarity calculated from MACCS descriptors. Hydrophobic – hydrophobic interaction. Metal acceptor – in this case the coordination bond between haem iron and heterocyclic nitrogen. "+": interaction present, "−": absent; bH: hydrophobic, MA: metal acceptor.
Molecular docking data can therefore be used to infer the structure–activity relationship. The substituent on the C-5 atom of tetrahydroimidazo[1,2-a]pyrimidine favors the inhibitory activity of this class of compounds, whereas the position of the substituent on the C-6 atom disrupts it. Furthermore, only the (S)-isomers of the compounds under consideration are expected to be potent inhibitors of CYP51.
The analysis of the interaction fingerprints (IFP) between the docking ligands and the protein shows that, similar to the reference ligand voriconazole, the compounds interact with the protein through hydrophobic interactions with hydrophobic residues of the protein and the formation of coordination bonds with the haem iron (Table 3). At the same time, hydrogen and ionic bonds are not observed for either the tested compounds or voriconazole. The Tanimoto coefficient, calculated for the interaction footprints of compounds 4a–e relative to voriconazole, indicates a range of values between 0.60 and 0.67. This suggests that the interaction patterns of these compounds differ by approximately four amino acids. The Tanimoto coefficient for compound 5e is significantly lower (≈0.42), indicating that the interaction pattern of compound 5e is significantly different from that of voriconazole, which may explain its low predicted affinity. The Tanimoto coefficient calculated from the MACCS descriptors for the selected compounds is <0.5, indicating that the chemical structure of compounds 4a–e and 5e is significantly different from voriconazole.
The three-dimensional position of the selected compounds in the active site of the enzyme is shown in Figure 5A for compound 4e. The heterocyclic nitrogen is indeed orientated towards the haem iron, with an N–Fe distance of 2.8 Å, which is slightly higher than the distance of 2.1 Å in the crystal complex with the inhibitor voriconazole. The benzene substituent on the C-6 atom is located in a hydrophobic pocket formed by residues Y122, L508, F504 and I373. The superposition of the selected and docked ligands reveals that compounds 4a–e are arranged in an identical manner, whereas the position of compound 5e, particularly tetrahydroimidazo[1,2-a]pyrimidine, is significantly disparate (Figure 5B).
![[1860-5397-20-236-5]](/bjoc/content/figures/1860-5397-20-236-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: (A) Position of the (S)-isomer of compound 4e in the active site of CYP51 after molecular dockinga. (B) Superposition of docked compounds 4a–e and 5e in a CYP51 ACb. aThe amino acid residues of the protein are colored green, compound 4e is colored gold and the haem is colored grey. The heterocyclic nitrogen of compound 5e appears to be orientated towards iron and is located 2.8 Å away from it, which may indicate the formation of a coordination bond; the benzene moiety is located in a hydrophobic pocket. bThe conformations of compounds 4a–e are identical, whereas the position of compound 5e (red) is significantly different.
Figure 5: (A) Position of the (S)-isomer of compound 4e in the active site of CYP51 after molecular dockinga....
Conclusion
Thus, we have proposed an efficient way to construct tetrahydroimidazo[1,2-a]pyrimidines without substituents at the 4 and 5-positions of the imidazole fragment by the reaction of 2-aminoimidazole with N-arylitaconimides and N-substituted maleimides. With the aid of DFT calculations, the most probable reaction path of the interaction was suggested. Based on the docking data, N-aryl(alkyl)-7-oxo-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidin-5-carboxamides as (S)-isomers were shown to be potent inhibitors of CYP51. These results allow us to consider these compounds as potential CYP51 inhibitor candidates for further in vitro and in vivo testing as antifungal prodrugs.
Experimental
General procedure for preparation of N-R-7-oxo-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidine-5-carboxamides 4a–i: A mixture of 2-aminoimidazole hemisulfate (1, 0.66 g, 5 mmol), N-substituted maleimides 2a–i (5 mmol), sodium acetate (0.82 g, 10 mmol), and iPrOH (10 mL) was boiled for 1 h. The resulting precipitate was filtered off, washed with water (2 × 5 mL) and recrystallized from a mixture of DMF/iPrOH 1:2. The title compounds were obtained as white (4a–e) or pink (4f–i) solids.
General procedure for the synthesis of N-aryl-2-(7-oxo-5,6,7,8-tetrahydroimidazo[1,2-a]pyrimidin-6-yl)acetamides 5a–e: A mixture of 2-aminoimidazole hemisulfate (1, 0.66 g, 5 mmol), N-arylitaconimide 3a–e (5 mmol), sodium acetate (0.82 g, 10 mmol), and iPrOH (10 mL) was boiled for 1 h. After cooling to rt, the resulting precipitate was filtered off, washed with water (2 × 5 mL) and recrystallized from a mixture of DMF/iPrOH 1:2. The title compounds 5a–e were obtained as white solids.
Supporting Information
| Supporting Information File 1: General reaction procedures, compound characterization data, copies of NMR and mass spectra for all new products. | ||
| Format: PDF | Size: 2.4 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Konovalov, A. I.; Antipin, I. S.; Burilov, V. A.; Madzhidov, T. I.; Kurbangalieva, A. R.; Nemtarev, A. V.; Solovieva, S. E.; Stoikov, I. I.; Mamedov, V. A.; Zakharova, L. Ya.; Gavrilova, E. L.; Sinyashin, O. G.; Balova, I. A.; Vasilyev, A. V.; Zenkevich, I. G.; Krasavin, M. Yu.; Kuznetsov, M. A.; Molchanov, A. P.; Novikov, M. S.; Nikolaev, V. A.; Rodina, L. L.; Khlebnikov, A. F.; Beletskaya, I. P.; Vatsadze, S. Z.; Gromov, S. P.; Zyk, N. V.; Lebedev, A. T.; Lemenovskii, D. A.; Petrosyan, V. S.; Nenaidenko, V. G.; Negrebetskii, V. V.; Baukov, Yu. I.; Shmigol’, T. A.; Korlyukov, A. A.; Tikhomirov, A. S.; Shchekotikhin, A. E.; Traven’, V. F.; Voskresenskii, L. G.; Zubkov, F. I.; Golubchikov, O. A.; Semeikin, A. S.; Berezin, D. B.; Stuzhin, P. A.; Filimonov, V. D.; Krasnokutskaya, E. A.; Fedorov, A. Yu.; Nyuchev, A. V.; Orlov, V. Yu.; Begunov, R. S.; Rusakov, A. I.; Kolobov, A. V.; Kofanov, E. R.; Fedotova, O. V.; Egorova, A. Yu.; Charushin, V. N.; Chupakhin, O. N.; Klimochkin, Yu. N.; Osyanin, V. A.; Reznikov, A. N.; Fisyuk, A. S.; Sagitullina, G. P.; Aksenov, A. V.; Aksenov, N. A.; Grachev, M. K.; Maslennikova, V. I.; Koroteev, M. P.; Brel’, A. K.; Lisina, S. V.; Medvedeva, S. M.; Shikhaliev, K. S.; Suboch, G. A.; Tovbis, M. S.; Mironovich, L. M.; Ivanov, S. M.; Kurbatov, S. V.; Kletskii, M. E.; Burov, O. N.; Kobrakov, K. I.; Kuznetsov, D. N. Russ. J. Org. Chem. 2018, 54, 157–371. doi:10.1134/s107042801802001x
Return to citation in text: [1] [2] -
Chern, J.-W.; Lee, C.-C.; Liaw, Y.-C.; Wang, A. H.-J. Heterocycles 1992, 34, 1133–1145. doi:10.3987/com-92-6000
Return to citation in text: [1] -
Chowdhury, M. G.; Kalmegh, V.; Kapoor, S.; Kamble, V.; Shard, A. RSC Med. Chem. 2024, 15, 1488–1507. doi:10.1039/d3md00718a
Return to citation in text: [1] -
Goel, R.; Luxami, V.; Paul, K. RSC Adv. 2015, 5, 81608–81637. doi:10.1039/c5ra14795f
Return to citation in text: [1] [2] -
Kobak, R. Z. U.; Akkurt, B. J. Turk. Chem. Soc., Sect. A 2022, 9, 1335–1386. doi:10.18596/jotcsa.1110922
Return to citation in text: [1] -
Azzouzi, M.; Ouafi, Z. E.; Azougagh, O.; Daoudi, W.; Ghazal, H.; Barkany, S. E.; Abderrazak, R.; Mazières, S.; Aatiaoui, A. E.; Oussaid, A. J. Mol. Struct. 2023, 1285, 135525. doi:10.1016/j.molstruc.2023.135525
Return to citation in text: [1] -
Feely, M.; Boyland, P.; Picardo, A.; Cox, A.; Gent, J. P. Eur. J. Pharmacol. 1989, 164, 377–380. doi:10.1016/0014-2999(89)90482-2
Return to citation in text: [1] -
Tully, W. R.; Gardner, C. R.; Gillespie, R. J.; Westwood, R. J. Med. Chem. 1991, 34, 2060–2067. doi:10.1021/jm00111a021
Return to citation in text: [1] -
Atack, J. R. Expert Opin. Invest. Drugs 2005, 14, 601–618. doi:10.1517/13543784.14.5.601
Return to citation in text: [1] -
Aeluri, R.; Alla, M.; Polepalli, S.; Jain, N. Eur. J. Med. Chem. 2015, 100, 18–23. doi:10.1016/j.ejmech.2015.05.037
Return to citation in text: [1] -
Rival, Y.; Grassy, G.; Taudou, A.; Ecalle, R. Eur. J. Med. Chem. 1991, 26, 13–18. doi:10.1016/0223-5234(91)90208-5
Return to citation in text: [1] -
Kishbaugh, T. L. S. Curr. Top. Med. Chem. 2016, 16, 3274–3302. doi:10.2174/1568026616666160506145141
Return to citation in text: [1] -
Cesur, Z.; Cesur, N.; Birteksöz, S.; Otük, G. Acta Chim. Slov. 2010, 57, 355–362.
Return to citation in text: [1] -
Kaplancikli, Z. A.; Turan-Zitouni, G.; Özdemr, A.; Revial, G. J. Enzyme Inhib. Med. Chem. 2008, 23, 866–870. doi:10.1080/14756360701811114
Return to citation in text: [1] -
Kethireddy, S.; Eppakayala, L.; Maringanti, T. C. Chem. Cent. J. 2015, 9, 51. doi:10.1186/s13065-015-0121-4
Return to citation in text: [1] -
El Kazzouli, S.; Berteina-Raboin, S.; Mouaddib, A.; Guillaumet, G. Tetrahedron Lett. 2003, 44, 6265–6267. doi:10.1016/s0040-4039(03)01532-6
Return to citation in text: [1] -
Cosimelli, B.; Laneri, S.; Ostacolo, C.; Sacchi, A.; Severi, E.; Porcù, E.; Rampazzo, E.; Moro, E.; Basso, G.; Viola, G. Eur. J. Med. Chem. 2014, 83, 45–56. doi:10.1016/j.ejmech.2014.05.071
Return to citation in text: [1] -
Ermolat'ev, D. S.; Giménez, V. N.; Babaev, E. V.; Van der Eycken, E. J. Comb. Chem. 2006, 8, 659–663. doi:10.1021/cc060031b
Return to citation in text: [1] -
Velázquez-Olvera, S.; Salgado-Zamora, H.; Velázquez-Ponce, M.; Campos-Aldrete, E.; Reyes-Arellano, A.; Pérez-González, C. Chem. Cent. J. 2012, 6, 83. doi:10.1186/1752-153x-6-83
Return to citation in text: [1] -
Li, H.; Zhao, J.; Zeng, L.; Hu, W. J. Org. Chem. 2011, 76, 8064–8069. doi:10.1021/jo201301p
Return to citation in text: [1] -
Li, J.; Zhou, G.; Hong, Y.; Wang, C.; He, W.; Wang, S.; Chen, Y.; Wen, Z.; Wang, Q. ACS Omega 2020, 5, 4868–4874. doi:10.1021/acsomega.9b03691
Return to citation in text: [1] -
Yang, S.; Thacker, Z.; Allison, E.; Bennett, M.; Cole, N.; Pinhero, P. J. ACS Appl. Mater. Interfaces 2017, 9, 40921–40929. doi:10.1021/acsami.7b04721
Return to citation in text: [1] -
Abdel‐Naby, A. S. J. Appl. Polym. Sci. 2011, 121, 169–175. doi:10.1002/app.33507
Return to citation in text: [1] -
Fleš, D.; Vuković, R.; Erceg Kuzmić, A.; Bogdanić, G.; Piližota, V.; Markuš, K.; Wolsperger, K.; Vikić-Topić, D. Croat. Chem. Acta 2023, 76, 69–74.
Return to citation in text: [1] -
Vandyshev, D. Yu.; Shikhaliev, K. S.; Kokonova, A. V.; Potapov, A. Yu.; Kolpakova, M. G.; Sabynin, A. L.; Zubkov, F. I. Chem. Heterocycl. Compd. 2016, 52, 493–497. doi:10.1007/s10593-016-1914-7
Return to citation in text: [1] [2] -
Vandyshev, D. Yu.; Shikhaliev, K. S.; Potapov, A. Yu.; Krysin, M. Yu.; Zubkov, F. I.; Sapronova, L. V. Beilstein J. Org. Chem. 2017, 13, 2561–2568. doi:10.3762/bjoc.13.252
Return to citation in text: [1] [2] -
Kovygin, Yu. A.; Shikhaliev, K. S.; Krysin, M. Yu.; Potapov, A. Yu.; Ledenyova, I. V.; Kosheleva, E. A.; Vandyshev, D. Yu. Chem. Heterocycl. Compd. 2019, 55, 748–754. doi:10.1007/s10593-019-02530-5
Return to citation in text: [1] [2] -
Vandyshev, D. Yu.; Shikhaliev, K. S. Molecules 2022, 27, 5268. doi:10.3390/molecules27165268
Return to citation in text: [1] [2] [3] [4] -
Siddharth, S.; Vittal, R. R. Arch. Microbiol. 2019, 201, 737–746. doi:10.1007/s00203-019-01634-y
Return to citation in text: [1] -
Bazine, I.; Bendjedid, S.; Boukhari, A. Arch. Pharm. (Weinheim, Ger.) 2021, 354, e2000291. doi:10.1002/ardp.202000291
Return to citation in text: [1] -
Raman, N.; Sakthivel, A.; Rajasekaran, K. Mycobiology 2007, 35, 150.
Return to citation in text: [1] -
Osmaniye, D.; Baltacı Bozkurt, N.; Levent, S.; Benli Yardımcı, G.; Sağlık, B. N.; Ozkay, Y.; Kaplancıklı, Z. A. ACS Omega 2023, 8, 24573–24585. doi:10.1021/acsomega.3c02797
Return to citation in text: [1] -
Dascalu, A.-E.; Ghinet, A.; Lipka, E.; Furman, C.; Rigo, B.; Fayeulle, A.; Billamboz, M. Fitoterapia 2020, 143, 104581. doi:10.1016/j.fitote.2020.104581
Return to citation in text: [1] -
Vandyshev, D. Yu.; Shikhaliev, K. S.; Potapov, A. Yu. Eur. Chem. Bull. 2015, 4, 424–427.
Return to citation in text: [1] -
Vandyshev, D. Yu.; Kovygin, Yu. A.; Khmelevskaya, T. N.; Sherbakov, K. A.; Shikhalieva, K. D.; Smoliannikova, M. Yu.; Shikhaliev, Kh. S. Russ. Chem. Bull. 2024, 73, 994–1004. doi:10.1007/s11172-024-4213-y
Return to citation in text: [1] -
Shmoylova, Y. Yu.; Kovygin, Y. A.; Kosheleva, E. A.; Shikhaliev, K. S.; Ledenyova, I. V.; Prezent, M. A. Mendeleev Commun. 2022, 32, 688–690. doi:10.1016/j.mencom.2022.09.041
Return to citation in text: [1] -
Rudenko, R. V.; Komykhov, S. A.; Musatov, V. I.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. J. Heterocycl. Chem. 2011, 48, 888–895. doi:10.1002/jhet.660
Return to citation in text: [1] -
Cuenca-Estrella, M.; Bernal-Martinez, L.; Buitrago, M. J.; Castelli, M. V.; Gomez-Lopez, A.; Zaragoza, O.; Rodriguez-Tudela, J. L. Int. J. Antimicrob. Agents 2008, 32 (Suppl. 2), 143–147. doi:10.1016/s0924-8579(08)70016-5
Return to citation in text: [1] -
Olaechea, P. M.; Palomar, M.; León-Gil, C.; Álvarez-Lerma, F.; Jordá, R.; Nolla-Salas, J.; León-Regidor, M. A.; EPCAN Study Group. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 323–330. doi:10.1007/s10096-004-1104-x
Return to citation in text: [1] -
Desai, J. V.; van de Veerdonk, F. L.; Lionakis, M. S. Intensive Care Med. 2018, 44, 1310–1314. doi:10.1007/s00134-017-4988-5
Return to citation in text: [1] -
Kullberg, B. J.; Arendrup, M. C. N. Engl. J. Med. 2015, 373, 1445–1456. doi:10.1056/nejmra1315399
Return to citation in text: [1] -
Tobudic, S.; Kratzer, C.; Presterl, E. Mycoses 2012, 55 (Suppl. 1), 24–32. doi:10.1111/j.1439-0507.2011.02146.x
Return to citation in text: [1] -
Rabelo, V. W.; Santos, T. F.; Terra, L.; Santana, M. V.; Castro, H. C.; Rodrigues, C. R.; Abreu, P. A. Fundam. Clin. Pharmacol. 2017, 31, 37–53. doi:10.1111/fcp.12230
Return to citation in text: [1] -
Rezaei, Z.; Khabnadideh, S.; Zomorodian, K.; Pakshir, K.; Kashi, G.; Sanagoei, N.; Gholami, S. Arch. Pharm. (Weinheim, Ger.) 2011, 344, 658–665. doi:10.1002/ardp.201000357
Return to citation in text: [1] -
Gonçalves, S. S.; Souza, A. C. R.; Chowdhary, A.; Meis, J. F.; Colombo, A. L. Mycoses 2016, 59, 198–219. doi:10.1111/myc.12469
Return to citation in text: [1] -
Masiá-Canuto, M.; Gutiérrez-Rodero, F. Lancet Infect. Dis. 2002, 2, 550–563. doi:10.1016/s1473-3099(02)00371-7
Return to citation in text: [1] -
Sanapalli, B. K. R.; Ashames, A.; Sigalapalli, D. K.; Shaik, A. B.; Bhandare, R. R.; Yele, V. Antibiotics (Basel, Switz.) 2022, 11, 1680. doi:10.3390/antibiotics11121680
Return to citation in text: [1] -
Rival, Y.; Taudou, A.; Ecalle, R. Farmaco 1993, 48, 857–869. doi:10.1002/chin.199509141
Return to citation in text: [1] -
Gómez-García, O.; Andrade-Pavón, D.; Campos-Aldrete, E.; Ballinas-Indilí, R.; Méndez-Tenorio, A.; Villa-Tanaca, L.; Álvarez-Toledano, C. Molecules 2018, 23, 599. doi:10.3390/molecules23030599
Return to citation in text: [1]
| 1. | Konovalov, A. I.; Antipin, I. S.; Burilov, V. A.; Madzhidov, T. I.; Kurbangalieva, A. R.; Nemtarev, A. V.; Solovieva, S. E.; Stoikov, I. I.; Mamedov, V. A.; Zakharova, L. Ya.; Gavrilova, E. L.; Sinyashin, O. G.; Balova, I. A.; Vasilyev, A. V.; Zenkevich, I. G.; Krasavin, M. Yu.; Kuznetsov, M. A.; Molchanov, A. P.; Novikov, M. S.; Nikolaev, V. A.; Rodina, L. L.; Khlebnikov, A. F.; Beletskaya, I. P.; Vatsadze, S. Z.; Gromov, S. P.; Zyk, N. V.; Lebedev, A. T.; Lemenovskii, D. A.; Petrosyan, V. S.; Nenaidenko, V. G.; Negrebetskii, V. V.; Baukov, Yu. I.; Shmigol’, T. A.; Korlyukov, A. A.; Tikhomirov, A. S.; Shchekotikhin, A. E.; Traven’, V. F.; Voskresenskii, L. G.; Zubkov, F. I.; Golubchikov, O. A.; Semeikin, A. S.; Berezin, D. B.; Stuzhin, P. A.; Filimonov, V. D.; Krasnokutskaya, E. A.; Fedorov, A. Yu.; Nyuchev, A. V.; Orlov, V. Yu.; Begunov, R. S.; Rusakov, A. I.; Kolobov, A. V.; Kofanov, E. R.; Fedotova, O. V.; Egorova, A. Yu.; Charushin, V. N.; Chupakhin, O. N.; Klimochkin, Yu. N.; Osyanin, V. A.; Reznikov, A. N.; Fisyuk, A. S.; Sagitullina, G. P.; Aksenov, A. V.; Aksenov, N. A.; Grachev, M. K.; Maslennikova, V. I.; Koroteev, M. P.; Brel’, A. K.; Lisina, S. V.; Medvedeva, S. M.; Shikhaliev, K. S.; Suboch, G. A.; Tovbis, M. S.; Mironovich, L. M.; Ivanov, S. M.; Kurbatov, S. V.; Kletskii, M. E.; Burov, O. N.; Kobrakov, K. I.; Kuznetsov, D. N. Russ. J. Org. Chem. 2018, 54, 157–371. doi:10.1134/s107042801802001x |
| 8. | Tully, W. R.; Gardner, C. R.; Gillespie, R. J.; Westwood, R. J. Med. Chem. 1991, 34, 2060–2067. doi:10.1021/jm00111a021 |
| 28. | Vandyshev, D. Yu.; Shikhaliev, K. S. Molecules 2022, 27, 5268. doi:10.3390/molecules27165268 |
| 29. | Siddharth, S.; Vittal, R. R. Arch. Microbiol. 2019, 201, 737–746. doi:10.1007/s00203-019-01634-y |
| 7. | Feely, M.; Boyland, P.; Picardo, A.; Cox, A.; Gent, J. P. Eur. J. Pharmacol. 1989, 164, 377–380. doi:10.1016/0014-2999(89)90482-2 |
| 30. | Bazine, I.; Bendjedid, S.; Boukhari, A. Arch. Pharm. (Weinheim, Ger.) 2021, 354, e2000291. doi:10.1002/ardp.202000291 |
| 31. | Raman, N.; Sakthivel, A.; Rajasekaran, K. Mycobiology 2007, 35, 150. |
| 32. | Osmaniye, D.; Baltacı Bozkurt, N.; Levent, S.; Benli Yardımcı, G.; Sağlık, B. N.; Ozkay, Y.; Kaplancıklı, Z. A. ACS Omega 2023, 8, 24573–24585. doi:10.1021/acsomega.3c02797 |
| 33. | Dascalu, A.-E.; Ghinet, A.; Lipka, E.; Furman, C.; Rigo, B.; Fayeulle, A.; Billamboz, M. Fitoterapia 2020, 143, 104581. doi:10.1016/j.fitote.2020.104581 |
| 4. | Goel, R.; Luxami, V.; Paul, K. RSC Adv. 2015, 5, 81608–81637. doi:10.1039/c5ra14795f |
| 5. | Kobak, R. Z. U.; Akkurt, B. J. Turk. Chem. Soc., Sect. A 2022, 9, 1335–1386. doi:10.18596/jotcsa.1110922 |
| 6. | Azzouzi, M.; Ouafi, Z. E.; Azougagh, O.; Daoudi, W.; Ghazal, H.; Barkany, S. E.; Abderrazak, R.; Mazières, S.; Aatiaoui, A. E.; Oussaid, A. J. Mol. Struct. 2023, 1285, 135525. doi:10.1016/j.molstruc.2023.135525 |
| 24. | Fleš, D.; Vuković, R.; Erceg Kuzmić, A.; Bogdanić, G.; Piližota, V.; Markuš, K.; Wolsperger, K.; Vikić-Topić, D. Croat. Chem. Acta 2023, 76, 69–74. |
| 2. | Chern, J.-W.; Lee, C.-C.; Liaw, Y.-C.; Wang, A. H.-J. Heterocycles 1992, 34, 1133–1145. doi:10.3987/com-92-6000 |
| 3. | Chowdhury, M. G.; Kalmegh, V.; Kapoor, S.; Kamble, V.; Shard, A. RSC Med. Chem. 2024, 15, 1488–1507. doi:10.1039/d3md00718a |
| 25. | Vandyshev, D. Yu.; Shikhaliev, K. S.; Kokonova, A. V.; Potapov, A. Yu.; Kolpakova, M. G.; Sabynin, A. L.; Zubkov, F. I. Chem. Heterocycl. Compd. 2016, 52, 493–497. doi:10.1007/s10593-016-1914-7 |
| 26. | Vandyshev, D. Yu.; Shikhaliev, K. S.; Potapov, A. Yu.; Krysin, M. Yu.; Zubkov, F. I.; Sapronova, L. V. Beilstein J. Org. Chem. 2017, 13, 2561–2568. doi:10.3762/bjoc.13.252 |
| 27. | Kovygin, Yu. A.; Shikhaliev, K. S.; Krysin, M. Yu.; Potapov, A. Yu.; Ledenyova, I. V.; Kosheleva, E. A.; Vandyshev, D. Yu. Chem. Heterocycl. Compd. 2019, 55, 748–754. doi:10.1007/s10593-019-02530-5 |
| 28. | Vandyshev, D. Yu.; Shikhaliev, K. S. Molecules 2022, 27, 5268. doi:10.3390/molecules27165268 |
| 4. | Goel, R.; Luxami, V.; Paul, K. RSC Adv. 2015, 5, 81608–81637. doi:10.1039/c5ra14795f |
| 21. | Li, J.; Zhou, G.; Hong, Y.; Wang, C.; He, W.; Wang, S.; Chen, Y.; Wen, Z.; Wang, Q. ACS Omega 2020, 5, 4868–4874. doi:10.1021/acsomega.9b03691 |
| 22. | Yang, S.; Thacker, Z.; Allison, E.; Bennett, M.; Cole, N.; Pinhero, P. J. ACS Appl. Mater. Interfaces 2017, 9, 40921–40929. doi:10.1021/acsami.7b04721 |
| 16. | El Kazzouli, S.; Berteina-Raboin, S.; Mouaddib, A.; Guillaumet, G. Tetrahedron Lett. 2003, 44, 6265–6267. doi:10.1016/s0040-4039(03)01532-6 |
| 17. | Cosimelli, B.; Laneri, S.; Ostacolo, C.; Sacchi, A.; Severi, E.; Porcù, E.; Rampazzo, E.; Moro, E.; Basso, G.; Viola, G. Eur. J. Med. Chem. 2014, 83, 45–56. doi:10.1016/j.ejmech.2014.05.071 |
| 18. | Ermolat'ev, D. S.; Giménez, V. N.; Babaev, E. V.; Van der Eycken, E. J. Comb. Chem. 2006, 8, 659–663. doi:10.1021/cc060031b |
| 19. | Velázquez-Olvera, S.; Salgado-Zamora, H.; Velázquez-Ponce, M.; Campos-Aldrete, E.; Reyes-Arellano, A.; Pérez-González, C. Chem. Cent. J. 2012, 6, 83. doi:10.1186/1752-153x-6-83 |
| 23. | Abdel‐Naby, A. S. J. Appl. Polym. Sci. 2011, 121, 169–175. doi:10.1002/app.33507 |
| 11. | Rival, Y.; Grassy, G.; Taudou, A.; Ecalle, R. Eur. J. Med. Chem. 1991, 26, 13–18. doi:10.1016/0223-5234(91)90208-5 |
| 12. | Kishbaugh, T. L. S. Curr. Top. Med. Chem. 2016, 16, 3274–3302. doi:10.2174/1568026616666160506145141 |
| 13. | Cesur, Z.; Cesur, N.; Birteksöz, S.; Otük, G. Acta Chim. Slov. 2010, 57, 355–362. |
| 14. | Kaplancikli, Z. A.; Turan-Zitouni, G.; Özdemr, A.; Revial, G. J. Enzyme Inhib. Med. Chem. 2008, 23, 866–870. doi:10.1080/14756360701811114 |
| 15. | Kethireddy, S.; Eppakayala, L.; Maringanti, T. C. Chem. Cent. J. 2015, 9, 51. doi:10.1186/s13065-015-0121-4 |
| 9. | Atack, J. R. Expert Opin. Invest. Drugs 2005, 14, 601–618. doi:10.1517/13543784.14.5.601 |
| 10. | Aeluri, R.; Alla, M.; Polepalli, S.; Jain, N. Eur. J. Med. Chem. 2015, 100, 18–23. doi:10.1016/j.ejmech.2015.05.037 |
| 20. | Li, H.; Zhao, J.; Zeng, L.; Hu, W. J. Org. Chem. 2011, 76, 8064–8069. doi:10.1021/jo201301p |
| 28. | Vandyshev, D. Yu.; Shikhaliev, K. S. Molecules 2022, 27, 5268. doi:10.3390/molecules27165268 |
| 1. | Konovalov, A. I.; Antipin, I. S.; Burilov, V. A.; Madzhidov, T. I.; Kurbangalieva, A. R.; Nemtarev, A. V.; Solovieva, S. E.; Stoikov, I. I.; Mamedov, V. A.; Zakharova, L. Ya.; Gavrilova, E. L.; Sinyashin, O. G.; Balova, I. A.; Vasilyev, A. V.; Zenkevich, I. G.; Krasavin, M. Yu.; Kuznetsov, M. A.; Molchanov, A. P.; Novikov, M. S.; Nikolaev, V. A.; Rodina, L. L.; Khlebnikov, A. F.; Beletskaya, I. P.; Vatsadze, S. Z.; Gromov, S. P.; Zyk, N. V.; Lebedev, A. T.; Lemenovskii, D. A.; Petrosyan, V. S.; Nenaidenko, V. G.; Negrebetskii, V. V.; Baukov, Yu. I.; Shmigol’, T. A.; Korlyukov, A. A.; Tikhomirov, A. S.; Shchekotikhin, A. E.; Traven’, V. F.; Voskresenskii, L. G.; Zubkov, F. I.; Golubchikov, O. A.; Semeikin, A. S.; Berezin, D. B.; Stuzhin, P. A.; Filimonov, V. D.; Krasnokutskaya, E. A.; Fedorov, A. Yu.; Nyuchev, A. V.; Orlov, V. Yu.; Begunov, R. S.; Rusakov, A. I.; Kolobov, A. V.; Kofanov, E. R.; Fedotova, O. V.; Egorova, A. Yu.; Charushin, V. N.; Chupakhin, O. N.; Klimochkin, Yu. N.; Osyanin, V. A.; Reznikov, A. N.; Fisyuk, A. S.; Sagitullina, G. P.; Aksenov, A. V.; Aksenov, N. A.; Grachev, M. K.; Maslennikova, V. I.; Koroteev, M. P.; Brel’, A. K.; Lisina, S. V.; Medvedeva, S. M.; Shikhaliev, K. S.; Suboch, G. A.; Tovbis, M. S.; Mironovich, L. M.; Ivanov, S. M.; Kurbatov, S. V.; Kletskii, M. E.; Burov, O. N.; Kobrakov, K. I.; Kuznetsov, D. N. Russ. J. Org. Chem. 2018, 54, 157–371. doi:10.1134/s107042801802001x |
| 25. | Vandyshev, D. Yu.; Shikhaliev, K. S.; Kokonova, A. V.; Potapov, A. Yu.; Kolpakova, M. G.; Sabynin, A. L.; Zubkov, F. I. Chem. Heterocycl. Compd. 2016, 52, 493–497. doi:10.1007/s10593-016-1914-7 |
| 26. | Vandyshev, D. Yu.; Shikhaliev, K. S.; Potapov, A. Yu.; Krysin, M. Yu.; Zubkov, F. I.; Sapronova, L. V. Beilstein J. Org. Chem. 2017, 13, 2561–2568. doi:10.3762/bjoc.13.252 |
| 27. | Kovygin, Yu. A.; Shikhaliev, K. S.; Krysin, M. Yu.; Potapov, A. Yu.; Ledenyova, I. V.; Kosheleva, E. A.; Vandyshev, D. Yu. Chem. Heterocycl. Compd. 2019, 55, 748–754. doi:10.1007/s10593-019-02530-5 |
| 28. | Vandyshev, D. Yu.; Shikhaliev, K. S. Molecules 2022, 27, 5268. doi:10.3390/molecules27165268 |
| 34. | Vandyshev, D. Yu.; Shikhaliev, K. S.; Potapov, A. Yu. Eur. Chem. Bull. 2015, 4, 424–427. |
| 35. | Vandyshev, D. Yu.; Kovygin, Yu. A.; Khmelevskaya, T. N.; Sherbakov, K. A.; Shikhalieva, K. D.; Smoliannikova, M. Yu.; Shikhaliev, Kh. S. Russ. Chem. Bull. 2024, 73, 994–1004. doi:10.1007/s11172-024-4213-y |
| 36. | Shmoylova, Y. Yu.; Kovygin, Y. A.; Kosheleva, E. A.; Shikhaliev, K. S.; Ledenyova, I. V.; Prezent, M. A. Mendeleev Commun. 2022, 32, 688–690. doi:10.1016/j.mencom.2022.09.041 |
| 47. | Sanapalli, B. K. R.; Ashames, A.; Sigalapalli, D. K.; Shaik, A. B.; Bhandare, R. R.; Yele, V. Antibiotics (Basel, Switz.) 2022, 11, 1680. doi:10.3390/antibiotics11121680 |
| 48. | Rival, Y.; Taudou, A.; Ecalle, R. Farmaco 1993, 48, 857–869. doi:10.1002/chin.199509141 |
| 49. | Gómez-García, O.; Andrade-Pavón, D.; Campos-Aldrete, E.; Ballinas-Indilí, R.; Méndez-Tenorio, A.; Villa-Tanaca, L.; Álvarez-Toledano, C. Molecules 2018, 23, 599. doi:10.3390/molecules23030599 |
| 43. | Rabelo, V. W.; Santos, T. F.; Terra, L.; Santana, M. V.; Castro, H. C.; Rodrigues, C. R.; Abreu, P. A. Fundam. Clin. Pharmacol. 2017, 31, 37–53. doi:10.1111/fcp.12230 |
| 44. | Rezaei, Z.; Khabnadideh, S.; Zomorodian, K.; Pakshir, K.; Kashi, G.; Sanagoei, N.; Gholami, S. Arch. Pharm. (Weinheim, Ger.) 2011, 344, 658–665. doi:10.1002/ardp.201000357 |
| 45. | Gonçalves, S. S.; Souza, A. C. R.; Chowdhary, A.; Meis, J. F.; Colombo, A. L. Mycoses 2016, 59, 198–219. doi:10.1111/myc.12469 |
| 46. | Masiá-Canuto, M.; Gutiérrez-Rodero, F. Lancet Infect. Dis. 2002, 2, 550–563. doi:10.1016/s1473-3099(02)00371-7 |
| 40. | Desai, J. V.; van de Veerdonk, F. L.; Lionakis, M. S. Intensive Care Med. 2018, 44, 1310–1314. doi:10.1007/s00134-017-4988-5 |
| 41. | Kullberg, B. J.; Arendrup, M. C. N. Engl. J. Med. 2015, 373, 1445–1456. doi:10.1056/nejmra1315399 |
| 42. | Tobudic, S.; Kratzer, C.; Presterl, E. Mycoses 2012, 55 (Suppl. 1), 24–32. doi:10.1111/j.1439-0507.2011.02146.x |
| 37. | Rudenko, R. V.; Komykhov, S. A.; Musatov, V. I.; Konovalova, I. S.; Shishkin, O. V.; Desenko, S. M. J. Heterocycl. Chem. 2011, 48, 888–895. doi:10.1002/jhet.660 |
| 38. | Cuenca-Estrella, M.; Bernal-Martinez, L.; Buitrago, M. J.; Castelli, M. V.; Gomez-Lopez, A.; Zaragoza, O.; Rodriguez-Tudela, J. L. Int. J. Antimicrob. Agents 2008, 32 (Suppl. 2), 143–147. doi:10.1016/s0924-8579(08)70016-5 |
| 39. | Olaechea, P. M.; Palomar, M.; León-Gil, C.; Álvarez-Lerma, F.; Jordá, R.; Nolla-Salas, J.; León-Regidor, M. A.; EPCAN Study Group. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 323–330. doi:10.1007/s10096-004-1104-x |
© 2024 Vandyshev et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.