Guest Editor: J. Wencel-Delord Beilstein J. Org. Chem.2025,21, 451–457.https://doi.org/10.3762/bjoc.21.32 Received 21 Nov 2024,
Accepted 12 Feb 2025,
Published 27 Feb 2025
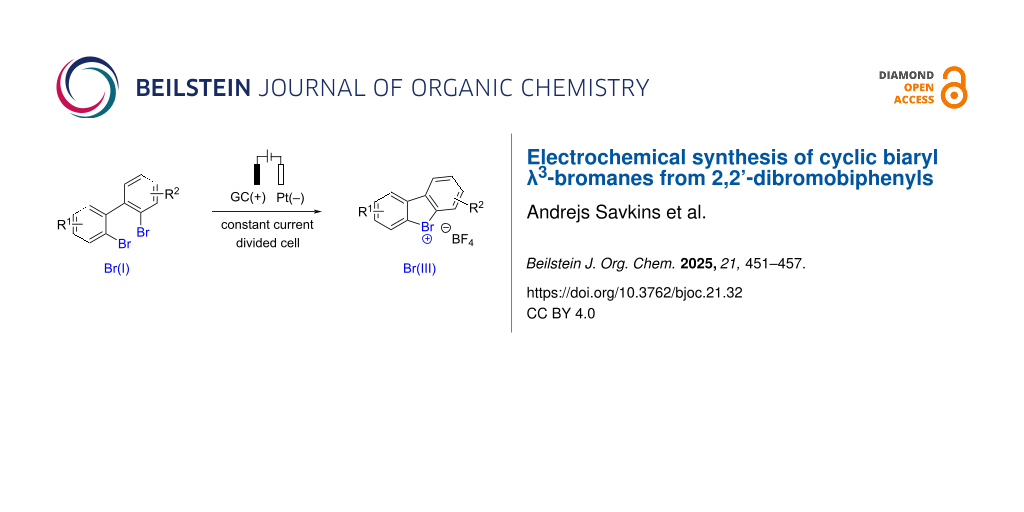
The remarkable nucleofugality of bromoarenes in diarylbromonium species renders them particularly suitable for the generation of arynes for subsequent use in a wide range of synthetic applications. The common approach to generate cyclic biaryl λ3-bromanes is based on thermal decomposition of hazardous diazonium salts. Herein, we disclose a mild and straightforward approach to diarylbromonium species by direct anodic oxidation of 2,2'-dibromo-1,1'-biphenyl. The electrochemical method provides access to a range of symmetrically and non-symmetrically substituted cyclic biaryl λ3-bromanes in moderate yields.
Chemistry of hypervalent bromine(III) species has experienced rapid advancements during the recent years [1,2]. The remarkable nucleofugality of aryl bromides in hypervalent bromine(III) compounds has been exploited in the generation of arynes from cyclic diaryl λ3-bromanes under remarkably mild conditions with subsequent applications of the in situ-generated arynes in cycloaddition reactions [3], meta-selective reactions with oxygen and nitrogen nucleophiles [4,5], regiodivergent meta or ortho-alkynylations [6], and regioselective (di)halogenation [7]. In addition, cyclic diaryl λ3-bromanes have been successfully employed as halogen-bonding organocatalysts in Michael addition [8] and their chiral variants were efficient in catalyzing enantioselective Mannich reactions of ketimines with cyanomethyl coumarins [9] and malonic esters [10]. These notable examples underscore the remarkable potential of cyclic diaryl λ3-bromanes in the development of efficient synthetic transformations.
Cyclic diaryl λ3-bromanes 1 are typically synthesized using a method developed by Sandin and Hay in 1952 [11] that relies on the excellent nucleofugality of molecular nitrogen in diazonium compounds. Accordingly, pre-formed [12] or in situ-generated 2,2’-bromodiazonium salts 2[11,13,14] furnish cyclic bromine(III) species 1 under thermal decomposition conditions (Scheme 1, reaction 1). The diazonium intermediates 2 are obtained by diazotation of 2'-bromo-[1,1'-biphenyl]-2-amines 3 with sodium nitrite and an acid under aqueous conditions. Recently, Wencel-Delord and co-workers disclosed an improved protocol toward diazonium intermediates 2 under non-aqueous conditions (t-BuONO and MsOH in acetonitrile) [4]. Nevertheless, the improved method still required thermal decomposition of the diazonium salt to effect the cyclization. In the quest for mild (room temperature) and scalable conditions toward cyclic diaryl λ3-bromanes 1 we realized that bromanyl units possess leaving group abilities comparable to the diazonium moiety [1,15]. Hence, the oxidation of 2,2'-dibromo-[1,1'-biphenyl] into mono-λ3-bromane 5 would set the stage for the key cyclization event (Scheme 1, reaction 2). We also reasoned that an anodic oxidation of the aryl bromide is perfectly suited for the generation of mono-λ3-bromane 5 under mild conditions [16-18]. This approach would conceptually differ from previously reported anodic syntheses of cyclic diaryl iodonium compounds, where an electrochemically generated acyclic iodine(III) intermediate undergoes an intramolecular SEAr-type reaction to form the cyclic product [19,20]. Herein, we report on the development of an electrochemical synthesis of cyclic diaryl λ3-bromanes under anodic oxidation conditions.
Scheme 1:
Synthesis of cyclic diarylbromonium compounds.
Scheme 1:
Synthesis of cyclic diarylbromonium compounds.
Symmetric 2,2'-dibromo-1,1'-biphenyl 4a possessing ethoxycarbonyl groups ortho to the bromine was chosen as a model compound for our study. We anticipated that the presence of the ester moiety would help to stabilize the key λ3-bromane(III) intermediate 5, as demonstrated in the work of Miyamoto et al. [21], thus facilitating its formation in anodic oxidation. The anodic oxidation of 4a under previously published conditions for the synthesis of Br(III) species [16,18] (GC as working electrode, Pt foil as counter electrode, 0.1 M tetrabutylammonium tetrafluoroborate (TBA-BF4) in HFIP electrolyte and 2 F passed charge at a current density of 10 mA·cm−2) afforded the desired Br(III) product 1a in poor 14% NMR yield (Table 1, entry 1). The cyclic diarylbromonium salt 1a was isolated by extractive workup followed by reversed-phase chromatography and its structure was unambiguously confirmed by X-ray crystallography (Table 1, graphic). Gratifyingly, a two-fold increase in the yield was achieved by a slight reduction of the current density to j = 8 mA·cm−2 (entry 2 vs entry 1 in Table 1). A further increase in the yield of 1a to 45% was possible by increasing the amount of passing charge (6.0 F; Table 1, entry 3). Tetraethylammonium tetrafluoroborate (TEA-BF4) appeared to be somewhat superior as the electrolyte to TBA-BF4 and tetramethylammonium tetrafluoroborate (TMA-BF4) (entry 4 vs entries 3 and 5, Table 1). In all experiments with a passed charge of 6.0 F (Table 1, entries 3–5), nearly complete conversion of starting 4a and moderate yield of the desired product 1a was observed pointing at a possible degradation of either starting material or product. Linear sweep voltammetry (LVS) experiments (0.1 M TBA-BF4 in HFIP on a Pt disk electrode) revealed that the reduction current increases almost 4 times upon the addition of 5 mM 1a to the electrolyte (see Supporting Information File 1, Figure S1). At the same time, passing 6.0 F through a solution of 1a in 50 mM TBA-BF4/HFIP at j = 8 mA·cm−2 led to 60% λ3-bromane 1a degradation, suggesting that cationic 1a, formed on the anode, decomposes on the cathode. To avoid the undesired cathodic decomposition of 1a, the cathode and anode chambers were separated, and further experiments were performed in a divided cell. Gratifyingly, the change of the cell type allowed for more than a two-fold increase in the yield of product 1a from 28% (entry 2, undivided cell, Table 1) to 60% (entry 6, divided cell). Interestingly, higher amounts of passed charge did not improve the reaction outcome (Table 1, entry 7). Neither successful was also a change of the anode material from GC to RVC or BDD (entries 8 and 9, Table 1), variation of electrolyte amount (entries 10 and 11) or altering of counter anions in the supporting electrolyte (entries 12 and 13; for complete optimization results, see Supporting Information File 1, Table S1).
Table 1:
Optimization of electrochemical oxidation/cyclization conditions.a
Entry
Deviation from starting conditions
1a [%]
4a [%]
Mass balance [%]
1
none
14
61
75
2
j = 8 mA cm−2
28
49
77
3
j = 8 mA cm−2, q/mol = 6.0 F
45
5
50
4
TEA-BF4, j = 8 mA cm−2, q/mol = 6.0 F
48
5
53
5
TMA -BF4, j = 8 mA cm−2, q/mol = 6.0 F
42
6
48
6
divided cell, 0.25 M TEA-BF4, j = 8 mA cm−2
60
24
84
7
divided cell, 0.25 M TEA-BF4, j = 8 mA cm−2, q/mol = 3.0 F
41
19
60
8
divided cell, RVC, 0.25 M TEA-BF4, j = 8 mA cm−2
62
0
62
9
divided cell, BDD, 0.25 M TEA-BF4, j = 8 mA cm−2
57
10
67
10
divided cell, 0.20 M TEA-BF4, j = 8 mA cm−2
15
55
70
11
divided cell, 0.30 M TEA-BF4, j = 8 mA cm−2
15
63
78
12
divided cell, 0.25 M TEA-ClO4, j = 8 mA cm−2
31
26
57
13
divided cell, 0.25 M TEA-PF6, j = 8 mA cm−2
46
19
65
aReactions were performed on a 0.15 mmol scale. Yields and mass balance were determined by 1H NMR in the crude reaction mixture using 1,2,3,4-tetrafluorobenzene as an internal standard. bEllipsoids are shown at 50% probability, with hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (deg) for 1a: Br5–O11, 2.657(2); Br5–O16, 2.647(2); Br5–C4A, 1.917(3); Br5–C5A, 1.915(2); Br5–F4, 3.081(5); C4A–Br5–C5A, 86.6(1); C4A–C9B–C9A-C5A, 1.0(3); C4A–C4–C10–O11, 0.1(4), and C5A–C6–C15–O16, 7.8(4).
With the optimized reaction conditions in hand, next, the substrate scope was evaluated (Scheme 2). Symmetrical biaryls with electron-deficient substituents such as Cl (4b) or CF3 (4c) afforded the respective Br(III) products 1b,c in slightly reduced yields as compared to that of 1a. Gratifyingly, electron-rich MeO-substituted 1d could be also obtained under the developed conditions. Unsymmetrically substituted 4e and monosubstituted dibromides 4f,g demonstrated reactivity similar to that of their symmetrical analogues 4b,c with exception of the mono-MeO-substituted dibromide 4h. Notably, the presence of two stabilizing ester moieties is critical for the synthesis of Br(III) species: the removal of one ester group (4i–k), or its replacement by NO2 (4l) or SO2t-Bu (4m) substituents in 2,2'-dibromo-1,1'-biphenyls resulted in starting material degradation with no formation of the desired product (for a complete list of substrates that do not form the desired electrochemical oxidation product see Supporting Information File 1, Scheme S1).
Scheme 2:
Substrate scope. Reactions were performed on a 0.15 mmol scale. Yields were determined by 1H NMR spectroscopy of the reaction mixture using 1,2,3,4-tetrafluorobenzene as an internal standard. Isolated yields are given in parentheses.
Scheme 2:
Substrate scope. Reactions were performed on a 0.15 mmol scale. Yields were determined by 1H NMR sp...
A series of control experiments was performed to rationalize the observed reactivity trends (Scheme 2). The measured redox potentials EP/2 for 2,2'-dibromo-1,1'-biphenyls 4a–g (from 1.77 V to 2.88 V) and unsuccessful substrates 4i–m, S9a, S9b (from 1.73 V to 2.54 V) (Table S2 in Supporting Information File 1) span similar potential regimes suggesting that the success of the anodic oxidation likely depends on the structural rather than the electrochemical properties of the starting 2,2'-dibromo-1,1'-biphenyls 4a–m. A closer inspection of the electrochemical behaviour of compound 4a revealed an irreversible electron transfer at scan rates up to 1 V s−1 (Scheme 3A) indicating that one or more rapid chemical steps are following the electrochemical step [22,23]. Besides, the observed linear correlation between Ep of the redox event and the square root of the scan rate (Scheme 3B) suggested that compound 4a is not significantly adsorbed on the electrode surface [23]. Comparison of the jp vs v0.5 slope with our previously obtained results for the anodic oxidation of aryl bromides 6a (two-electron oxidation) and 6b (one-electron oxidation) into the respective bromine(III) species (Scheme 3C) [17] demonstrated a more similar behaviour to 6b suggesting that revealed oxidation is a single-electron-transfer process. It is important to note that this comparison assumes that the diffusion coefficients of 4a and 6, parameters that influence the jp vs v0.5 slope, are similar. On the other hand, anodic oxidation of 4a under optimized conditions (entry 6, Table 1) using 1 F returned only 35% of 1a (NMR yield), whereas passing 2 F charge delivered 1a in 60% yield. The latter data provides an evidence that the overall oxidation of 4a to 1a likely is a two-electron process, suggesting that the second oxidation step may involve disproportionation of putative Br(II) species (see Scheme 3D).
Scheme 3:
A: Background and iR drop-corrected CVs of 5 mM 4a at different scan rates (solvent: HFIP, working electrode: glassy carbon, supporting electrolyte: 0.1 M TBA-BF4). B: Plot of the peak current densities (jp) vs v0.5. C: Representative jp vs v0.5 slope values for oxidation of Martin’s bromane precursor 6 (ref. [17]). D: Plausible reaction mechanism.
Scheme 3:
A: Background and iR drop-corrected CVs of 5 mM 4a at different scan rates (solvent: HFIP, working ...
Based on the control experiments described above we propose a plausible mechanism as shown in Scheme 3D. The reaction starts with a single-electron oxidation of 4a on the electrode surface to form cation radical A, in which Br(II) is chelation-stabilized by the carboxyl group [21] and the neighbouring Br substituent [24]. Intermediate A rapidly undergoes irreversible chemical reaction by HFIP coordination to transient Br(II) followed by subsequent deprotonation to generate radical B. A following disproportionation of radical B would lead to the formation of Br(III) species C (anodic oxidation cannot be fully excluded), which undergoes intramolecular SNAr-type substitution to form cyclic λ3-bromane 1a and hypobromite D. The latter decomposes into bromide and hexafluoroacetone, with the latter observed by 19F NMR [25]. Assuming that intermediate B is sufficiently stable to leave the diffusion layer and undergoes disproportionation to the species C in the bulk electrolyte provides a reasonable explanation for why the reaction appears as a one-electron oxidation in CV experiments, but still as a two-electron oxidation in electrolysis.
Conclusion
In conclusion, we have demonstrated a conceptual approach to cyclic diaryl λ3-bromanes by electrochemical oxidative cyclization of 2,2'-dibromo-1,1'-biphenyls. The developed method represents a safe and inexpensive alternative to the commonly used thermal decomposition of potentially explosive diazonium salts. The successful electrochemical oxidation requires the presence of two chelating ester groups that stabilize the formed Br(III) species. Further work towards improving the substrate scope and understanding the reaction mechanism are in progress in our laboratory.
Supporting Information
Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre and allocated the deposition number CCDC 2404146.
Supporting Information File 1:
Experimental procedures, analytical and spectroscopic data for new compounds, copies of NMR spectra, and X-ray crystallographic data.
The authors thank Dr. Sergey Belyakov from the Latvian Institute of Organic Synthesis (LIOS) for X-ray crystallographic analysis. We also thank Prof. Edgars Suna (LIOS) for helpful discussions and assistance in preparing the manuscript.
Funding
This work was financially supported by Latvian Science Council grant LZP-2021/1-0595.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
Lanzi, M.; Wencel-Delord, J. Chem. Sci.2024,15, 1557–1569. doi:10.1039/d3sc05382b
Return to citation in text:
[1]
[2]
Winterson, B.; Patra, T.; Wirth, T. Synthesis2022,54, 1261–1271. doi:10.1055/a-1675-8404
Return to citation in text:
[1]
Lanzi, M.; Ali Abdine, R. A.; De Abreu, M.; Wencel-Delord, J. Org. Lett.2021,23, 9047–9052. doi:10.1021/acs.orglett.1c03278
Return to citation in text:
[1]
Lanzi, M.; Dherbassy, Q.; Wencel‐Delord, J. Angew. Chem., Int. Ed.2021,60, 14852–14857. doi:10.1002/anie.202103625
Return to citation in text:
[1]
[2]
Wang, Y.; Tian, Y.-N.; Ren, S.; Zhu, R.; Huang, B.; Wen, Y.; Li, S. Org. Chem. Front.2023,10, 793–798. doi:10.1039/d2qo01406h
Return to citation in text:
[1]
De Abreu, M.; Rogge, T.; Lanzi, M.; Saiegh, T. J.; Houk, K. N.; Wencel-Delord, J. Angew. Chem., Int. Ed.2024,63, e202319960. doi:10.1002/anie.202319960
Return to citation in text:
[1]
Carter Martos, D.; de Abreu, M.; Hauk, P.; Fackler, P.; Wencel-Delord, J. Chem. Sci.2024,15, 6770–6776. doi:10.1039/d4sc01234h
Return to citation in text:
[1]
Yoshida, Y.; Ishikawa, S.; Mino, T.; Sakamoto, M. Chem. Commun.2021,57, 2519–2522. doi:10.1039/d0cc07733j
Return to citation in text:
[1]
Yoshida, Y.; Mino, T.; Sakamoto, M. ACS Catal.2021,11, 13028–13033. doi:10.1021/acscatal.1c04070
Return to citation in text:
[1]
Yoshida, Y.; Ao, T.; Mino, T.; Sakamoto, M. Molecules2023,28, 384. doi:10.3390/molecules28010384
Return to citation in text:
[1]
Sandin, R. B.; Hay, A. S. J. Am. Chem. Soc.1952,74, 274–275. doi:10.1021/ja01121a524
Return to citation in text:
[1]
[2]
Riedmüller, S.; Nachtsheim, B. J. Beilstein J. Org. Chem.2013,9, 1202–1209. doi:10.3762/bjoc.9.136
Return to citation in text:
[1]
Huss, C. D.; Yoshimura, A.; Rohde, G. T.; Mironova, I. A.; Postnikov, P. S.; Yusubov, M. S.; Saito, A.; Zhdankin, V. V. ACS Omega2024,9, 2664–2673. doi:10.1021/acsomega.3c07512
Return to citation in text:
[1]
Nakajima, M.; Miyamoto, K.; Hirano, K.; Uchiyama, M. J. Am. Chem. Soc.2019,141, 6499–6503. doi:10.1021/jacs.9b02436
Return to citation in text:
[1]
Sokolovs, I.; Mohebbati, N.; Francke, R.; Suna, E. Angew. Chem., Int. Ed.2021,60, 15832–15837. doi:10.1002/anie.202104677
Return to citation in text:
[1]
[2]
Mohebbati, N.; Sokolovs, I.; Woite, P.; Lõkov, M.; Parman, E.; Ugandi, M.; Leito, I.; Roemelt, M.; Suna, E.; Francke, R. Chem. – Eur. J.2022,28, e202200974. doi:10.1002/chem.202200974
Return to citation in text:
[1]
[2]
[3]
Scherkus, A.; Gudkova, A.; Čada, J.; Müller, B. H.; Bystron, T.; Francke, R. J. Org. Chem.2024,89, 14129–14134. doi:10.1021/acs.joc.4c01521
Return to citation in text:
[1]
Elsherbini, M.; Moran, W. J. Org. Biomol. Chem.2021,19, 4706–4711. doi:10.1039/d1ob00457c
Return to citation in text:
[1]
Miyamoto, K.; Saito, M.; Tsuji, S.; Takagi, T.; Shiro, M.; Uchiyama, M.; Ochiai, M. J. Am. Chem. Soc.2021,143, 9327–9331. doi:10.1021/jacs.1c04536
Return to citation in text:
[1]
[2]
Heinze, J. Angew. Chem., Int. Ed. Engl.1984,23, 831–847. doi:10.1002/anie.198408313
Return to citation in text:
[1]
Sandford, C.; Edwards, M. A.; Klunder, K. J.; Hickey, D. P.; Li, M.; Barman, K.; Sigman, M. S.; White, H. S.; Minteer, S. D. Chem. Sci.2019,10, 6404–6422. doi:10.1039/c9sc01545k
Return to citation in text:
[1]
[2]
Frey, B. L.; Figgins, M. T.; Van Trieste, G. P., III; Carmieli, R.; Powers, D. C. J. Am. Chem. Soc.2022,144, 13913–13919. doi:10.1021/jacs.2c05562
Return to citation in text:
[1]
Anderson, J. D. O.; DesMarteau, D. D. J. Fluorine Chem.1996,77, 147–152. doi:10.1016/0022-1139(96)03400-8
Return to citation in text:
[1]
Reference 23
23.
Sandford, C.; Edwards, M. A.; Klunder, K. J.; Hickey, D. P.; Li, M.; Barman, K.; Sigman, M. S.; White, H. S.; Minteer, S. D. Chem. Sci.2019,10, 6404–6422. doi:10.1039/c9sc01545k
Sandford, C.; Edwards, M. A.; Klunder, K. J.; Hickey, D. P.; Li, M.; Barman, K.; Sigman, M. S.; White, H. S.; Minteer, S. D. Chem. Sci.2019,10, 6404–6422. doi:10.1039/c9sc01545k
De Abreu, M.; Rogge, T.; Lanzi, M.; Saiegh, T. J.; Houk, K. N.; Wencel-Delord, J. Angew. Chem., Int. Ed.2024,63, e202319960. doi:10.1002/anie.202319960
Riedmüller, S.; Nachtsheim, B. J. Beilstein J. Org. Chem.2013,9, 1202–1209. doi:10.3762/bjoc.9.136
14.
Huss, C. D.; Yoshimura, A.; Rohde, G. T.; Mironova, I. A.; Postnikov, P. S.; Yusubov, M. S.; Saito, A.; Zhdankin, V. V. ACS Omega2024,9, 2664–2673. doi:10.1021/acsomega.3c07512



![[1860-5397-21-32-i1]](/bjoc/content/inline/1860-5397-21-32-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-32-i4.svg?max-width=637&scale=1.0)
![[1860-5397-21-32-i2]](/bjoc/content/inline/1860-5397-21-32-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-21-32-i3]](/bjoc/content/inline/1860-5397-21-32-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)