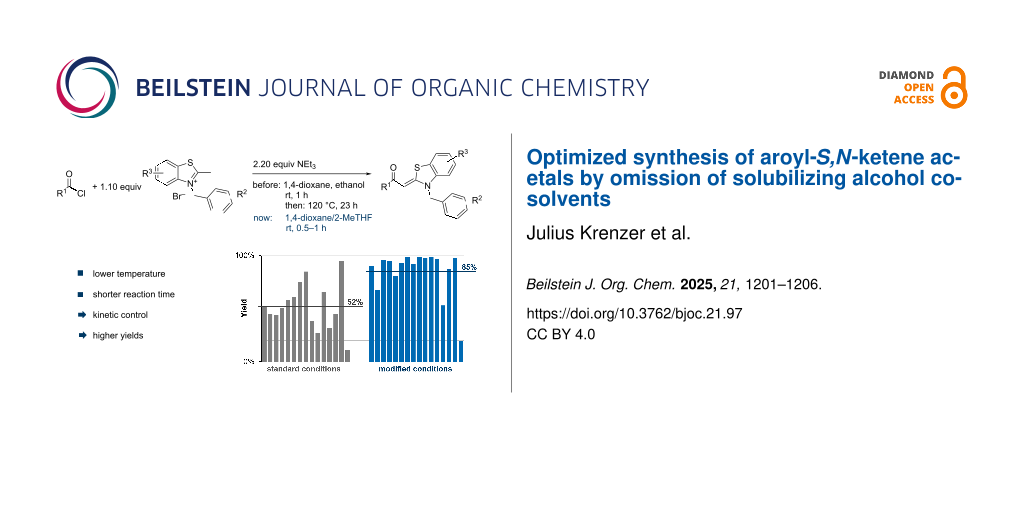
Abstract
Aroyl-S,N-ketene acetals are obtained by condensation of aroyl chlorides and 2-methyl-N-benzylbenzothiazolium salts in 1,4-dioxane at room temperature in short reaction time in 20–99% yield. This protocol represents a considerable improvement over the standard synthesis in 1,4-dioxane/ethanol mixtures at elevated temperatures.

Graphical Abstract
Introduction
Aroyl-S,N-ketene acetals, in particular N-benzyl derivatives 1, are very short donor–acceptor chromophores that have recently found a renaissance due to their peculiar intense solid-state emission and significant turn-on of emission upon induced aggregation in alcohol–water mixtures [1]. This chromophore class has been extensively developed in recent years, even to aggregation-induced emissive (AIE) multichromophores [2] and even bichromophoric fluorimetric sensors [3,4]. The retrosynthesis of the title compounds 1 suggests starting from aroyl chlorides 2 and 2-methyl-N-benzylbenzothiazolium salts 3 by a condensation transform (Scheme 1). The condensation essentially represents an addition–elimination sequence that starts with the nucleophilic S,N-ketene acetal 4, in situ generated from substrate 3 by deprotonation, followed by chloride elimination/neutralization from the zwitterionic tetrahedral intermediate 5 to give the target molecule 1.
![[1860-5397-21-97-i1]](/bjoc/content/inline/1860-5397-21-97-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of aroyl-S,N-ketene acetals 1 and tentative mechanistic scenario of the addition–elimination sequence.
Scheme 1: Retrosynthetic analysis of aroyl-S,N-ketene acetals 1 and tentative mechanistic scenario of the add...
The standard protocol for the synthesis of (hetero)aroyl-S,N-ketene acetals 8 from (hetero)aroyl chlorides 6 and 2-methylbenzothiazolium salts 7 employs a twofold excess of an amine base in a binary 1,4-dioxane/ethanol mixture. Ethanol was used as a cosolvent to ensure solubility of the polar intermediate according to the mechanistic rationale (Scheme 2) [5,6]. Although, a broad scope of diversely substituted (hetero)aroyl-S,N-ketene acetals 8 was obtained (111 examples), the average yield of 57% indicates that the process might require optimization, in particular, for further methodological implementation. Here, we report on the improved synthesis of (hetero)aroyl-S,N-ketene acetals 8 by careful solvent and temperature optimization.
![[1860-5397-21-97-i2]](/bjoc/content/inline/1860-5397-21-97-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Standard protocol for the synthesis of (hetero)aroyl-S,N-ketene acetals 8 in binary dioxane/ethanol mixtures.
Scheme 2: Standard protocol for the synthesis of (hetero)aroyl-S,N-ketene acetals 8 in binary dioxane/ethanol...
Results and Discussion
The presumed byproducts in the addition–elimination sequence in the presence of an excess of ethanol as a cosolvent are the ethyl ester formed by Einhorn acylation [7] of the acid chloride under the standard conditions and deep colored polar byproducts (according to TLC detection) that arise from self-condensation of 2-methylbenzothiazolium salts 7 and intermediary formed S,N-ketene acetals 4 at elevated temperatures. Einhorn acylation is governed by the electrophilicity of the acid chloride or acylammonium species and the nucleophilicity of any nucleophilic species present in the reaction mixture. In addition, since condensations are affected by the bimolecular addition as an elementary step, special attention to the concentration of the nucleophiles has to be paid.
According to Mayr’s nucleophilicity scales [8-10], a nucleophilicity parameter N for the specific S,N-ketene acetal intermediate 4 can estimated from parameters for enamines (N = 10–16), cyclic N,N-ketene acetals (N = 18–20) [11,12] , and the deoxy-Breslow intermediate (N =15.6) [13]. The nucleophilicity of S,N-ketene acetal 4 clearly exceeds that of ethanol (N = 7.4). In addition, triethylamine is more nucleophilic (N = 17.30 in dichloromethane; N = 17.10 in acetonitrile) [14] than ethanol or the S,N-ketene acetal intermediate 4. Hence, mechanistically the acid chloride first transforms to an acylammonium species as in Einhorn acylations [7]. As a consequence, for avoiding any competing ethyl ester formation, ethanol has to be omitted from in the process. For suppressing the formation of side products by self-condensation of S,N-ketene acetal intermediates the reaction temperature has to be kept as low as possible for assuring kinetic control.
Therefore, in contrast to the standard protocol [5,6] (Scheme 2), reacting aroyl chlorides 2 and 2-methyl-N-benzylbenzothiazolium salts 3 in 1,4-dioxane as a solvent at room temp for 0.5 to 1 h produces representative aroyl-S,N-ketene acetals 1 in mostly excellent yield (Scheme 3). For comparison, the yields of the products 1 under standard (A) and new modified (B) conditions are listed in Table 1. The yield of compounds 1 after flash chromatography on silica gel under modified conditions (B) are significantly higher than under standard conditions (A).
![[1860-5397-21-97-i3]](/bjoc/content/inline/1860-5397-21-97-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Modified protocol for the synthesis of aroyl-S,N-ketene acetals 1 in dioxane at room temperature.
Scheme 3: Modified protocol for the synthesis of aroyl-S,N-ketene acetals 1 in dioxane at room temperature.
Table 1: Comparison of the yields of the condensation synthesis of aroyl-S,N-ketene acetals 1 under standard (A)a and modified conditions Bb and Cc.
| entry | acid chloride 2 | benzothiazolium salt 3 | aroyl-S,N-ketene acetal 1d |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-97-i4.svg?max-width=637&scale=1.0)
2a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-97-i5.svg?max-width=637&scale=1.0)
3a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-21-97-i6.svg?max-width=637&scale=1.0)
1a (A: 52% [5]; B: 90%) |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-21-97-i7.svg?max-width=637&scale=1.0)
2a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-21-97-i8.svg?max-width=637&scale=1.0)
3b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-21-97-i9.svg?max-width=637&scale=1.0)
1b (B: 68%) |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-21-97-i10.svg?max-width=637&scale=1.0)
2b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-21-97-i11.svg?max-width=637&scale=1.0)
3a |
![[Graphic 9]](/bjoc/content/inline/1860-5397-21-97-i12.svg?max-width=637&scale=1.0)
1c (A: 45% [6]; B: 96%) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-21-97-i13.svg?max-width=637&scale=1.0)
2c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-21-97-i14.svg?max-width=637&scale=1.0)
3a |
![[Graphic 12]](/bjoc/content/inline/1860-5397-21-97-i15.svg?max-width=637&scale=1.0)
1d (A: 44% [6]; B: 95%) |
| 5 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-21-97-i16.svg?max-width=637&scale=1.0)
2c |
![[Graphic 14]](/bjoc/content/inline/1860-5397-21-97-i17.svg?max-width=637&scale=1.0)
3b |
![[Graphic 15]](/bjoc/content/inline/1860-5397-21-97-i18.svg?max-width=637&scale=1.0)
1e (A: 51%; B: 81%) |
| 6 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-21-97-i19.svg?max-width=637&scale=1.0)
2d |
![[Graphic 17]](/bjoc/content/inline/1860-5397-21-97-i20.svg?max-width=637&scale=1.0)
3a |
![[Graphic 18]](/bjoc/content/inline/1860-5397-21-97-i21.svg?max-width=637&scale=1.0)
1f (A: 58% [5]; B: 93%; C: 95%) |
| 7 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-21-97-i22.svg?max-width=637&scale=1.0)
2d |
![[Graphic 20]](/bjoc/content/inline/1860-5397-21-97-i23.svg?max-width=637&scale=1.0)
3b |
![[Graphic 21]](/bjoc/content/inline/1860-5397-21-97-i24.svg?max-width=637&scale=1.0)
1g (A: 61% [6]; B: 99%) |
| 8e |
![[Graphic 22]](/bjoc/content/inline/1860-5397-21-97-i25.svg?max-width=637&scale=1.0)
2d |
![[Graphic 23]](/bjoc/content/inline/1860-5397-21-97-i26.svg?max-width=637&scale=1.0)
3d |
![[Graphic 24]](/bjoc/content/inline/1860-5397-21-97-i27.svg?max-width=637&scale=1.0)
1h (A: 75% [6]; B: 92%; C: 91%) |
| 9 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-21-97-i28.svg?max-width=637&scale=1.0)
2e |
![[Graphic 26]](/bjoc/content/inline/1860-5397-21-97-i29.svg?max-width=637&scale=1.0)
3a |
![[Graphic 27]](/bjoc/content/inline/1860-5397-21-97-i30.svg?max-width=637&scale=1.0)
1i (A: 85% [5]; B: 99%) |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-21-97-i31.svg?max-width=637&scale=1.0)
2f |
![[Graphic 29]](/bjoc/content/inline/1860-5397-21-97-i32.svg?max-width=637&scale=1.0)
3a |
![[Graphic 30]](/bjoc/content/inline/1860-5397-21-97-i33.svg?max-width=637&scale=1.0)
1j (A: 38% [6]; B: 98%) |
| 11 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-21-97-i34.svg?max-width=637&scale=1.0)
2g |
![[Graphic 32]](/bjoc/content/inline/1860-5397-21-97-i35.svg?max-width=637&scale=1.0)
3a |
![[Graphic 33]](/bjoc/content/inline/1860-5397-21-97-i36.svg?max-width=637&scale=1.0)
1k (A: 27% [6]; B: 99%) |
| 12 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-21-97-i37.svg?max-width=637&scale=1.0)
2h |
![[Graphic 35]](/bjoc/content/inline/1860-5397-21-97-i38.svg?max-width=637&scale=1.0)
3a |
![[Graphic 36]](/bjoc/content/inline/1860-5397-21-97-i39.svg?max-width=637&scale=1.0)
1l (A: 66% [5]; B: 97%) |
| 13 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-21-97-i40.svg?max-width=637&scale=1.0)
2h |
![[Graphic 38]](/bjoc/content/inline/1860-5397-21-97-i41.svg?max-width=637&scale=1.0)
3b |
![[Graphic 39]](/bjoc/content/inline/1860-5397-21-97-i42.svg?max-width=637&scale=1.0)
1m (A: 32% [6]; B: 53%) |
| 14 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-21-97-i43.svg?max-width=637&scale=1.0)
2h |
![[Graphic 41]](/bjoc/content/inline/1860-5397-21-97-i44.svg?max-width=637&scale=1.0)
3c |
![[Graphic 42]](/bjoc/content/inline/1860-5397-21-97-i45.svg?max-width=637&scale=1.0)
1n (A: 45%; B: 87%) |
| 15 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-21-97-i46.svg?max-width=637&scale=1.0)
2i |
![[Graphic 44]](/bjoc/content/inline/1860-5397-21-97-i47.svg?max-width=637&scale=1.0)
3a |
![[Graphic 45]](/bjoc/content/inline/1860-5397-21-97-i48.svg?max-width=637&scale=1.0)
1o (A: 95% [5]; B: 98%) |
| 16 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-21-97-i49.svg?max-width=637&scale=1.0)
2j |
![[Graphic 47]](/bjoc/content/inline/1860-5397-21-97-i50.svg?max-width=637&scale=1.0)
3a |
![[Graphic 48]](/bjoc/content/inline/1860-5397-21-97-i51.svg?max-width=637&scale=1.0)
1p (A: 11% [6]; B: 20%) |
aConditions A: Aroyl chloride 2 (1.0 equiv), 2-methyl-N-benzylbenzothiazolium salts 3 (1.1 equiv), and triethylamine (2.2 equiv) in a mixture of 1,4-dioxane and ethanol (34 equiv) were stirred at room temperature for 1 h and then at 120 °C for 23 h. bConditions B: Aroyl chloride 2 (1.0 equiv), 2-methyl-N-benzylbenzothiazolium salts 3 (1.1 equiv), and triethylamine (2.2 equiv) in 1,4-dioxane were stirred at room temperature for 0.5 to 1 h. cConditions C: Aroyl chloride 2 (1.0 equiv), 2-methyl-N-benzylbenzothiazolium salts 3 (1.1 equiv), and triethylamine (2.2 equiv) in 2-Me-THF were stirred at room temperature for 1 h. dYields after chromatography on silica gel. eA dilution to 8 mL of 1,4-dioxane per mmol was used.
The results demonstrate the broad applicability of the optimized reaction conditions. Acid chlorides with electron-withdrawing (-CN) and electron-donating (-OMe) substituents were used. Additionally, a heterocycle could be incorporated. Different benzyl substituents were employed, and substitution at the benzothiazole core was also tolerated (Table 1).
As an alternative to 1,4-dioxane, 2-methyltetrahydrofuran was tested the solvent. This can be obtained from renewable biomass and is therefore considered to be a "green" solvent. In 2-MeTHF (conditions (C)), product 1f was isolated with a yield of 95% and derivative 1h with a yield of 91%. This shows that the sustainable solvent is a potent alternative to 1,4-dioxane. With the new protocol, not only does the synthesis need less energy, it can also be carried out more sustainably, making it "greener."
Conclusion
Changing the solvent system from 1,4-dioxane/ethanol to 1,4-dioxane and reacting the aroyl chlorides and 2-methyl-N-benzylbenzothiazolium salts at room temp for 0.5–1 h gives rise to the formation of aroyl-S,N-ketene acetals in mostly excellent yield. The modification of this condensation synthesis is conceptualized on the basis of relative reactivities of intermediary formed acylammonium salts (electrophilicity) and S,N-ketene acetals (nucleophilicity). In addition, kinetic control is warranted by conducting the condensation synthesis at room temperature for only short reaction times. Furthermore, 2-MeTHF was successfully implemented as a sustainable alternative to 1,4-dioxane. This modified protocol is clearly superior to the initial protocol (in 1,4-dioxane/ethanol) and will be applied in future sequences for the generation of (hetero)aroyl-S,N-ketene acetals.
Experimental
Synthesis of compound 1i according to conditions B (typical procedure): 4-Fluorobenzoyl chloride (2e, 0.590 mL, 4.99 mmol, 1.00 equiv) and benzothiazolium salt 3a (1.76 g, 5.50 mmol, 1.10 equiv) were placed in a sintered, dry screw-cap Schlenk-tube under nitrogen atmosphere and dissolved in dry 1,4-dioxane (30 mL). Triethylamine (1.52 mL, 11.0 mmol, 2.20 equiv) was added to the reaction mixture and the solution was stirred for 1 h at room temperature. The crude product was absorbed onto Celite® and purified by flash chromatography on silica gel (n-hexane/acetone 3:1). Then, the crude product was suspended in n-hexane, the supernatant separated by filtration and the precipitate was dried under vacuum to afford aroyl-S,N-ketene acetal 1i (1.78 g, 4.93 mmol, 99%) as a yellow solid. Mp 159 °C (lit. 155 °C [5]). 1H NMR (300 MHz, CDCl3) δ 5.24 (s, 2H), 6.43 (s, 1H), 6.92–7.04 (m, 3H), 7.09–7.16 (m, 3H), 7.19–7.29 (m, 4H), 7.57 (dd, 3J = 7.7 Hz, 4J = 1.3 Hz, 1H), 7.73–7.83 (m, 2H); MALDI–TOF–MS (m/z): 362.2, [C22H16FNOS + H]+.
Supporting Information
| Supporting Information File 1: Experimental details of the synthesis and analytical data of compounds 1 and 3, 1H and 13C NMR spectra of compounds 1 and 3. | ||
| Format: PDF | Size: 1.5 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Biesen, L.; Müller, T. J. J. Chem. – Eur. J. 2023, 29, e202302067. doi:10.1002/chem.202302067
Return to citation in text: [1] -
Biesen, L.; Krenzer, J.; Nirmalananthan-Budau, N.; Resch-Genger, U.; Müller, T. J. J. Chem. Sci. 2022, 13, 5374–5381. doi:10.1039/d2sc00415a
Return to citation in text: [1] -
Biesen, L.; May, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Chem. – Eur. J. 2021, 27, 13426–13434. doi:10.1002/chem.202102052
Return to citation in text: [1] -
Biesen, L.; Müller, T. J. J. Aggregate 2021, 2, e105. doi:10.1002/agt2.105
Return to citation in text: [1] -
Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Wang, Z. Einhorn Acylation. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp 967–970. doi:10.1002/9780470638859.conrr209
Return to citation in text: [1] [2] -
Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c
Return to citation in text: [1] -
Mayr, H.; Ofial, A. R. Pure Appl. Chem. 2005, 77, 1807–1821. doi:10.1351/pac200577111807
Return to citation in text: [1] -
Mayr, H.; Ofial, A. R. J. Phys. Org. Chem. 2008, 21, 584–595. doi:10.1002/poc.1325
Return to citation in text: [1] -
Li, Z.; Ji, P.; Cheng, J.-P. J. Org. Chem. 2021, 86, 2974–2985. doi:10.1021/acs.joc.0c02838
Return to citation in text: [1] -
Maji, B.; Horn, M.; Mayr, H. Angew. Chem., Int. Ed. 2012, 51, 6231–6235. doi:10.1002/anie.201202327
Return to citation in text: [1] -
Maji, B.; Mayr, H. Angew. Chem., Int. Ed. 2012, 51, 10408–10412. doi:10.1002/anie.201204524
Return to citation in text: [1] -
Ammer, J.; Baidya, M.; Kobayashi, S.; Mayr, H. J. Phys. Org. Chem. 2010, 23, 1029–1035. doi:10.1002/poc.1707
Return to citation in text: [1]
| 1. | Biesen, L.; Müller, T. J. J. Chem. – Eur. J. 2023, 29, e202302067. doi:10.1002/chem.202302067 |
| 7. | Wang, Z. Einhorn Acylation. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp 967–970. doi:10.1002/9780470638859.conrr209 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 3. | Biesen, L.; May, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Chem. – Eur. J. 2021, 27, 13426–13434. doi:10.1002/chem.202102052 |
| 4. | Biesen, L.; Müller, T. J. J. Aggregate 2021, 2, e105. doi:10.1002/agt2.105 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 2. | Biesen, L.; Krenzer, J.; Nirmalananthan-Budau, N.; Resch-Genger, U.; Müller, T. J. J. Chem. Sci. 2022, 13, 5374–5381. doi:10.1039/d2sc00415a |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 14. | Ammer, J.; Baidya, M.; Kobayashi, S.; Mayr, H. J. Phys. Org. Chem. 2010, 23, 1029–1035. doi:10.1002/poc.1707 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 13. | Maji, B.; Mayr, H. Angew. Chem., Int. Ed. 2012, 51, 10408–10412. doi:10.1002/anie.201204524 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 11. | Li, Z.; Ji, P.; Cheng, J.-P. J. Org. Chem. 2021, 86, 2974–2985. doi:10.1021/acs.joc.0c02838 |
| 12. | Maji, B.; Horn, M.; Mayr, H. Angew. Chem., Int. Ed. 2012, 51, 6231–6235. doi:10.1002/anie.201202327 |
| 8. | Mayr, H.; Kempf, B.; Ofial, A. R. Acc. Chem. Res. 2003, 36, 66–77. doi:10.1021/ar020094c |
| 9. | Mayr, H.; Ofial, A. R. Pure Appl. Chem. 2005, 77, 1807–1821. doi:10.1351/pac200577111807 |
| 10. | Mayr, H.; Ofial, A. R. J. Phys. Org. Chem. 2008, 21, 584–595. doi:10.1002/poc.1325 |
| 7. | Wang, Z. Einhorn Acylation. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp 967–970. doi:10.1002/9780470638859.conrr209 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
| 6. | Biesen, L.; Woschko, D.; Janiak, C.; Müller, T. J. J. Chem. – Eur. J. 2022, 28, e202202579. doi:10.1002/chem.202202579 |
| 5. | Biesen, L.; Nirmalananthan‐Budau, N.; Hoffmann, K.; Resch‐Genger, U.; Müller, T. J. J. Angew. Chem., Int. Ed. 2020, 59, 10037–10041. doi:10.1002/anie.201916396 |
© 2025 Krenzer and Müller; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.