Abstract
Katsumadain A, a naturally occurring influenza virus neuraminidase (NA) inhibitor, was synthesized by using a bioinspired, organocatalytic enantioselective 1,4-conjugate addition of styryl-2-pyranone with cinnamaldehyde, followed by a tandem Horner–Wadsworth–Emmons/oxa Michael addition.

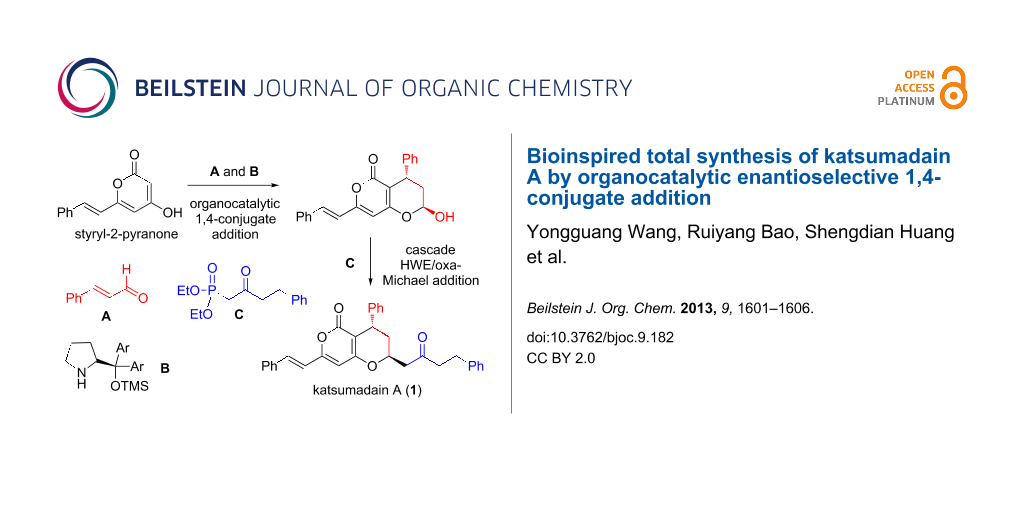
Graphical Abstract
Introduction
2-Pyranone is a privilege structure that is often present in natural products and pharmaceuticals, many of which exhibit diverse molecular architectures and biological profiles [1,2]. For example, katsumadain A (1) and B (2), which were isolated from Alpinia katsumadai Hayata (Zingiberaceae), a chinese herbal drug used as an anti-emetic and stomachic agent, are two natural products bearing a diarylheptanoid scaffold that is incorporated into the styryl-2-pyranone moiety [3]. Preliminary biological evaluations showed that 1 and 2 feature anti-emetic activities on copper sulfate-induced emesis in young chicks. More recently, Rollinger et al. disclosed that katsumadain A (1) exhibited prominent in vitro inhibitory activity against the human influenza virus A/PR/8/34 of the subtype H1N1 (IC50 1.05–0.42 μM) by targeting the enzyme neuraminidase (NA) [4,5]. Moreover, it also inhibited the NA of four H1N1 swine influenza viruses with IC50 values between 0.59 and 1.64 μM. Therefore, katsumadain A represents an attractive lead structure for the anti-flu drug discovery [6].
We recently reported the biomimetic total synthesis of katsumadain C [7], a natural product isolated from the same resource as katsumadain A and B [8]. As part of our continuous interest in the synthesis of bioactive 2-pyranone-derived natural products, we launched a project aiming to develop a highly efficient route for the synthesis of katasumadain A as well as its analogues, which would pave the way for their application in further biomedical investigations.
Biosynthetically, katsumadain A is assumed to be derived from styryl-2-pyranone 3 and alnustone (4) [9] through a 1,6-conjugate addition/oxa-Michael addition cascade reaction (path a, Scheme 1). Indeed, both 3 and 4 are known natural substances. Apparently, the biosynthetic pathway represents the most straightforward and convergent approach to synthesize katsumadain A. However, its efficiency might be limited to some extent, given that α,β,γ,δ-unsaturated ketone 4 could undergo a competitive 1,4-conjugate addition to provide the other natural product katsumadain B (path b, Scheme 1). Actually, the regioselectivity of a conjugate addition with α,β,γ,δ-unsaturated Michael acceptors remains a considerable challenge, as it is heavily dependent on the steric and electronic nature of the substrates [10,11]. Moreover, the enantioselective 1,6-conjugate addition to acyclic dienones or dienoates monosubstituted at the β- and δ-position has rarely been investigated [12-15], thus leaving open the question of whether or not a biomimetic approach towards katsumadain A might succeed. Keeping these concerns in mind, an alternative strategy was designed as a fallback, in which katsumadain A could be accessed from the lactol 5a and phosphonate 6 via a tandem Horner–Wadsworth–Emmons (HWE)/oxa-Michael addition reaction [16]. In turn, 5a could be derived from 3 and cinnamaldehyde (7) by an organocatalytic enantioselective 1,4-conjugate addition followed by the hemiketal formation.
![[1860-5397-9-182-i1]](/bjoc/content/inline/1860-5397-9-182-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed biosynthetic pathway and strategic analysis for synthesis of katsumadain A.
Scheme 1: Proposed biosynthetic pathway and strategic analysis for synthesis of katsumadain A.
Results and Discussion
Our investigation was initiated by an investigation of the conditions that could effect the proposed biomimetic approach towards katsumadain A and katsumadain B. Both styryl-2-pyranone 3 [17] and alnustone (4) [18] were synthesized according to the literature methods. First of all, the 1,6-conjugate addition of 3 towards 4 was attempted by employing various conditions, including different basic conditions (NaH, DBU, KHMDS) by activation of the nucleophile 3 or acidic conditions (AcOH, TMSOTf, Sc(OTf)3 and In(OTf)3) by activation of the electrophile 4. However, all of these reactions failed to provide satisfactory results and only lead to the recovery or the substantial decomposition of the starting material. We then turned our attention to the organocatalytic conjugated addition reaction. Among the various documented conditions [19-23], the 9-amino-9-deoxyepicinchona alkaloid-promoted Michael addition is particularly attractive, mainly due to the availability of the catalyst and its superior reactivity towards the activation of the unsaturated ketone substrates through formation of the corresponding iminium intermediate [22]. To our delight, when we tried the standard conditions (30% catalyst A, 60% TFA, DCM, 96 h) in our case, we isolated a product in 25% isolated yield, which was proved to be the 1,4-adduct katsumadain B (2). Encouraged by this result, we further optimized the reaction by screening different solvents (CH3CN, THF, DMSO and MeOH) and additives (HCl, TFA and DMAP), aiming to improve the efficiency and selectivity (1,4- or 1,6-adduct) of the reaction. In most of the cases the 1,4-conjugate addition proceeded dominantly, while no or only trace amounts of the 1,6-adduct katsumadain A (1) was observed. The best result was obtained when the reaction was performed with a substoichiometric amount of catalyst A with MeOH as a solvent, in which katsumadain A and B were isolated in a 5:6 ratio with a combined yield of 33% (Scheme 2).
![[1860-5397-9-182-i2]](/bjoc/content/inline/1860-5397-9-182-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preliminary results of the biomimetic synthesis of katsumadain A.
Scheme 2: Preliminary results of the biomimetic synthesis of katsumadain A.
With limited success regarding the the biomimetic synthesis of katsumadain A, we then moved towards the alternative approach as described in Scheme 1. We envisioned that in this scenario an organocatalytic 1,4-conjugate addition [24-27] between 3 and 7 would circumvent both the reactivity and selectivity issues, which we have struggled with in the aforementioned studies. To validate this hypothesis, we performed a systematic investigation of the organocatalytic 1,4-conjugate addition by examining various reaction parameters, including organocatalyst, acid additive, solvent temperature, and reaction temperature (Table 1). The first reaction was performed by stirring a mixture of 3 and 7 in DCM at room temperature for 12 h in the presence of Hayashi catalyst B [28]. It was found that the desired product 5a was obtained, albeit in moderate yield and enantioselectivity (Table 1, entry 1). To our delight, the usage of benzoic acid (BA) as an additive could dramatically improve the reaction by affording 5a in a good yield (78%) and a good ee value (91%, Table 1, entry 2). Besides the catalyst B, both Jørgensen catalyst C [29] and MacMillan catalyst D [30] were also tested in this reaction, but gave inferior results (Table 1, entries 3 and 4). As to the acid additive, p-nitrobenzoic acid (PNBA) was found to afford 5a in an excellent yield, but with a decreased ee value (80%). Furthermore, the solvent effect was also examined. Among the several solvents examined, both MeOH and DMSO proved to be suitable solvent systems (Table 1, entries 6 and 8) by furnishing comparable results with DCM, while MeCN and toluene led to modest results (Table 1, entry 7 and 9). Finally, we found that the reaction temperature has some influence on the outcomes, with a slightly improved enantioselectivity (92% ee) obtained at 0 °C (Table 1, entry 10 and 11). Although the best ee value (93%) was achieved at −20 °C, the reaction became sluggish and the yield dropped to 45% (Table 1, entry 12). It is noteworthy that 5a was isolated as a mixture of C-5 diastereoisomers (β-isomer/α-isomer = 5:1 to 7:1) in all of the above cases. For convenience, the ee value of 5a was determined with the corresponding lactone derivative. Furthermore, the absolute stereochemistry of 5a (β-isomer) was assigned as (7S,5R) by using the Mosher ester method (see Supporting Information File 1 for details).
Table 1: Condition screening of organocatalytic 1,4-conjugate addition/hemiketalization of styryl-2-pyranone with α,β-unsaturated aldehydes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-182-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | Catalyst | Additive | Solvent | T (°C) | Yield (%) | ee (%)b |
|---|---|---|---|---|---|---|
| 1 | B | none | DCM | 23 | 41 | 78 |
| 2 | B | BA | DCM | 23 | 78 | 91 |
| 3 | C | BA | DCM | 23 | 41 | −81 |
| 4 | D | BA | DCM | 23 | 10 | nd |
| 5 | B | PNBA | DCM | 23 | 94 | 80 |
| 6 | B | BA | MeOH | 23 | 80 | 91 |
| 7 | B | BA | CH3CN | 23 | 78 | 75 |
| 8 | B | BA | DMSO | 23 | 79 | 93 |
| 9 | B | BA | Toluene | 23 | 66 | 91 |
| 10 | B | BA | MeOH | 0 | 78 | 92 |
| 11 | B | BA | DCM | 0 | 82 | 92 |
| 12 | B | BA | DCM | −20 | 45 | 93 |
aEach reaction was run with 3 (0.5 mmol) and 7 (0.6 mmol) in 2.0 mL solvent as shown above. bThe ee value was measured with the corresponding lactone product of 5a using chiral HPLC.
To evaluate the substrate scope of the reaction, we then examined different substituted styryl-2-pyranone and cinnamaldehyde derivatives as Michael addtion donors and acceptors (Table 2). When styryl-2-pyranone 3a remained unchanged, a variety of cinnamaldehyde derivatives (7a–f) bearing either electron-withdrawing groups (4-Cl, 4-CF3 and 4-NO2, Table 2, entries 2-4) or electron-donating groups (4-MeO or 3,5-MeO, Table 2, entries 5 and 6) on the phenyl ring proved to be suitable substrates, affording the corresponding products (5b–f) in good yields and enantioselectivities. Besides 3a, the Michael addition donors could also be extended to other substituted styryl-2-pyranone derivatives (e.g., 3b and 3c, Table 2, entries 7–11), all of which gave acceptable results. As proof-of-concept cases, the above outcomes indicate that the developed organocatalytic enantioselective 1,4-conjugate addition could be potentially applied to the synthesis of various bicyclic compounds bearing different aromatic moieties (Ar1 and Ar2), which paves the way to access katsumadain A and its analogues for further biomedical studies.
Table 2: Substrate scope of organocatalytic 1,4-conjugate addition/hemiketalization of styryl-2-pyranones with α,β-unsaturated aldehydes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-182-i5.svg?max-width=637&scale=1.0)
|
|||
| Entrya | Substrate (3 and 7) | Yield of 5 (%)b | ee value of 5 (%)c |
|---|---|---|---|
| 1 | 3a: Ar1 = Ph; 7a: Ar2 = Ph | 82 (5a) | 92 |
| 2 | 3a: Ar1 = Ph; 7b: Ar2 = 4-Cl-Ph | 59 (5b) | 83 |
| 3 | 3a: Ar1 = Ph; 7c: Ar2 = 4-CF3-Ph | 77 (5c) | 82 |
| 4 | 3a: Ar1 = Ph; 7d: Ar2 = 4-NO2-Ph | 79 (5d) | 88 |
| 5 | 3a: Ar1 = Ph; 7e: Ar2 = 4-MeO-Ph | 78 (5e) | 87 |
| 6 | 3a: Ar1 = Ph; 7f: Ar2 = 3,5-MeO-Ph | 70 (5f) | 92 |
| 7 | 3b: Ar1 = 4-MeO-Ph; 7a: Ar2 = Ph | 84 (5g) | 82 |
| 8 | 3c: Ar1 = Furan; 7b: Ar2 = 4-Cl-Ph | 88 (5h) | 84 |
| 9 | 3c: Ar1 = Furan; 7d: Ar2 = 4-NO2-Ph | 76 (5i) | 91 |
| 10 | 3c: Ar1 = Furan; 7e: Ar2 = 4-MeO-Ph | 75 (5j) | 80 |
| 11 | 3c: Ar1 = Furan; 7g: Ar2 = Naphthyl | 79 (5k) | 90 |
aEach reaction was run with 3 (0.5 mmol) and 7 (0.6 mmol) in 2.0 mL solvent as shown above. bEach of 5a–k was obtained as a mixture of C-5 diastereoisomers (the ratio of α-isomer:β-isomer varied from 1:5 to 1:7. cThe ee value of 5a–k was measured with the corresponding lactone product using chiral HPLC.
Having achieved the bicyclic core of katsumadain A in an efficient and enantioselective manner, we then moved towards its total synthesis through the proposed tandem Horner–Wadsworth–Emmons/oxa-Michael addition. As expected, deprotonation of 6 [31] with KHMDS at −40 °C for 0.5 h followed by the addition of the lactol 5a led to the formation of katsumadain A as the only diastereoisomer in 52% yield, apparently via the in situ generated intermediate 8. The spectroscopic data of the synthetic katsumadain A were in accordance with those of the natural one [32]. However, we found that its optical rotation ([a]D25 = −75.4, c 0.40, EtOH) was quite different from the reported one ([a]D25 +3.7, c 0.40, EtOH), indicating that the naturally occurring 1 might exist as a racemic substance. Given that the two enantiomers of katsumadain A might show different behaviors in the biological studies from each other as well as from the racemic compounds, we also synthesized (+)-katsumadain A in a similar way by simply replacing the catatalyst B with its enantiomer in the organocatalytic 1,4-conjugate addition (Scheme 3).
![[1860-5397-9-182-i3]](/bjoc/content/inline/1860-5397-9-182-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Total synthesis of both enantiomers of katsumadain A.
Scheme 3: Total synthesis of both enantiomers of katsumadain A.
Conclusion
We accomplished the first enantioselective total synthesis of katsumadain A, a naturally occurring influenza virus neuraminidase (NA) inhibitor. The key elements of the synthesis featured a bioinspired, organocatalytic enantioselective 1,4-conjuate addition and a tandem HWE/oxa-Michael addition. Due to the high efficiency and flexibility of the synthetic route it is applicable to the syntheses of both enantiomers of katsumadain A as well as their analogues. Applications of these compounds in relevant biomedical studies are ongoing in this laboratory, and the progress will be reported in due course.
Experimental
Representative procedure for the organocatalytic 1,4-conjugate addition: To a mixture of 3a (214 mg, 1.0 mmol) and 7a (163 mg, 1.2 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added PhCOOH (24 mg, 0.2 mmol) and catatalyst B (50 mg, 0.2 equiv). The mixture was stirred at 0 °C for 10 h before being quenched by saturated aqueous NH4Cl. The mixture was extracted with DCM (3 × 10 mL), and the organic layers were washed with brine and dried over MgSO4. The organic solvent was removed under vacuum, and the residue was purified by column chromatography (CH2Cl2:ethyl acetate = 20:1) to give 5a (284 mg, 82% yield) as a light yellow solid.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data for synthetic 1, 3a–c, 5a–k and 9a–k. | ||
| Format: PDF | Size: 5.2 MB | Download |
Acknowledgements
This work was financially supported by the National Science Foundation of China (21102081, 21272133), the Beijing Natural Science Foundation (2132037), the New Teachers' Fund for Doctor Stations (20110002120011), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, and the Ministry of Education.
References
-
McGlacken, G. P.; Fairlamb, I. J. S. Nat. Prod. Rep. 2005, 22, 369–385. doi:10.1039/b416651p
Return to citation in text: [1] -
Goel, A.; Ram, V. J. Tetrahedron 2009, 65, 7865–7913. doi:10.1016/j.tet.2009.06.031
Return to citation in text: [1] -
Yang, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tai, T.; Nunoura, Y.; Watanabe, K. J. Nat. Prod. 1999, 62, 1672–1674. doi:10.1021/np990096e
Return to citation in text: [1] -
Grienke, U.; Schmidtke, M.; Kirchmair, J.; Pfarr, K.; Wutzler, P.; Dürrwald, R.; Wolber, G.; Liedl, K. R.; Stuppner, H.; Rollinger, J. M. J. Med. Chem. 2010, 53, 778–786. doi:10.1021/jm901440f
Return to citation in text: [1] -
Kirchmair, J.; Rollinger, J. M.; Liedl, K. R.; Seidel, N.; Krumbholz, A.; Schmidtke, M. Future Med. Chem. 2011, 3, 437–450. doi:10.4155/fmc.10.292
Return to citation in text: [1] -
Grienke, U.; Schmidtke, M.; von Grafenstein, S.; Kirchmair, J.; Liedl, K. R.; Rollinger, J. M. Nat. Prod. Rep. 2012, 29, 11–36. doi:10.1039/c1np00053e
Return to citation in text: [1] -
Zhang, P.; Wang, Y.; Bao, R.; Luo, T.; Yang, Z.; Tang, Y. Org. Lett. 2012, 14, 162–165. doi:10.1021/ol2029433
Return to citation in text: [1] -
Yang, C.-S.; Wang, X.-B.; Wang, J.-S.; Luo, J.-G.; Luo, L.; Kong, L.-Y. Org. Lett. 2011, 13, 3380–3383. doi:10.1021/ol201137v
Return to citation in text: [1] -
Kuroyanagi, M.; Noro, T.; Fukushima, S.; Aiyama, R.; Ikuta, A.; Itokawa, H.; Morita, M. Chem. Pharm. Bull. 1983, 31, 1544–1550. doi:10.1248/cpb.31.1544
Return to citation in text: [1] -
Csákÿ, A. G.; de la Herrán, G.; Murcia, M. C. Chem. Soc. Rev. 2010, 39, 4080–4102. doi:10.1039/b924486g
Return to citation in text: [1] -
Silva, E. M. P.; Silva, A. M. S. Synthesis 2012, 44, 3109–3128. doi:10.1055/s-0032-1316778
Return to citation in text: [1] -
den Hartog, T.; Harutyunyan, S. R.; Font, D.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 398–401. doi:10.1002/anie.200703702
Return to citation in text: [1] -
Bernardi, L.; López-Cantarero, J.; Niess, B.; Jørgensen, K. A. J. Am. Chem. Soc. 2007, 129, 5772–5778. doi:10.1021/ja0707097
Return to citation in text: [1] -
Murphy, J. J.; Quintard, A.; McArdle, P.; Alexakis, A.; Stephens, J. C. Angew. Chem., Int. Ed. 2011, 50, 5095–5098. doi:10.1002/anie.201100804
Return to citation in text: [1] -
Tian, X.; Liu, Y.; Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 6439–6442. doi:10.1002/anie.201202392
Return to citation in text: [1] -
McLeod, M. C.; Wilson, Z. E.; Brimble, M. A. Org. Lett. 2011, 13, 5382–5385. doi:10.1021/ol202265g
Return to citation in text: [1] -
Katritzky, A. R.; Wang, Z.; Wang, M.; Hall, C. D.; Suzuki, K. J. Org. Chem. 2005, 70, 4854–4856. doi:10.1021/jo050307m
Return to citation in text: [1] -
Baranovsky, A.; Schmitt, B.; Fowler, D. J.; Schneider, B. Synth. Commun. 2003, 33, 1019–1045. doi:10.1081/SCC-120016367
Return to citation in text: [1] -
Wang, J.-J.; Hu, Z.-P.; Lou, C.-L.; Liu, J.-L.; Li, X.-M.; Yan, M. Tetrahedron 2011, 67, 4578–4583. doi:10.1016/j.tet.2011.04.086
Return to citation in text: [1] -
Zhu, X.; Lin, A.; Shi, Y.; Guo, J.; Zhu, C.; Cheng, Y. Org. Lett. 2011, 13, 4382–4385. doi:10.1021/ol201715h
Return to citation in text: [1] -
Li, X.-M.; Wang, B.; Zhang, J.-M.; Yan, M. Org. Lett. 2011, 13, 374–377. doi:10.1021/ol102570b
Return to citation in text: [1] -
Xie, J.-W.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.-G.; Chen, Y.-C. Org. Lett. 2007, 9, 413–415. doi:10.1021/ol062718a
Return to citation in text: [1] [2] -
Liu, Y.; Liu, X.; Wang, M.; He, P.; Lin, L.; Feng, X. J. Org. Chem. 2012, 77, 4136–4142. doi:10.1021/jo3001047
Return to citation in text: [1] -
Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p
Return to citation in text: [1] -
Almasi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Rueping, M.; Merino, E.; Sugiono, E. Adv. Synth. Catal. 2008, 350, 2127–2131. doi:10.1002/adsc.200800340
Return to citation in text: [1] -
Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599
Return to citation in text: [1] -
Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 794–797. doi:10.1002/anie.200462101
Return to citation in text: [1] -
Austin, J. F.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 1172–1173. doi:10.1021/ja017255c
Return to citation in text: [1] -
Maloney, K. M.; Chung, J. Y. L. J. Org. Chem. 2009, 74, 7574–7576. doi:10.1021/jo901552k
Return to citation in text: [1] -
The ee value of the synthetic (+)-1and (−)-1 was determined to be 80% and 81%, respectively, suggesting that partial racemization occurred in the HWE reaction, presumably via the intermediate 8.
Return to citation in text: [1]
| 31. | Maloney, K. M.; Chung, J. Y. L. J. Org. Chem. 2009, 74, 7574–7576. doi:10.1021/jo901552k |
| 29. | Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 794–797. doi:10.1002/anie.200462101 |
| 30. | Austin, J. F.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 1172–1173. doi:10.1021/ja017255c |
| 1. | McGlacken, G. P.; Fairlamb, I. J. S. Nat. Prod. Rep. 2005, 22, 369–385. doi:10.1039/b416651p |
| 2. | Goel, A.; Ram, V. J. Tetrahedron 2009, 65, 7865–7913. doi:10.1016/j.tet.2009.06.031 |
| 7. | Zhang, P.; Wang, Y.; Bao, R.; Luo, T.; Yang, Z.; Tang, Y. Org. Lett. 2012, 14, 162–165. doi:10.1021/ol2029433 |
| 24. | Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p |
| 25. | Almasi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 26. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 27. | Rueping, M.; Merino, E.; Sugiono, E. Adv. Synth. Catal. 2008, 350, 2127–2131. doi:10.1002/adsc.200800340 |
| 6. | Grienke, U.; Schmidtke, M.; von Grafenstein, S.; Kirchmair, J.; Liedl, K. R.; Rollinger, J. M. Nat. Prod. Rep. 2012, 29, 11–36. doi:10.1039/c1np00053e |
| 28. | Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599 |
| 4. | Grienke, U.; Schmidtke, M.; Kirchmair, J.; Pfarr, K.; Wutzler, P.; Dürrwald, R.; Wolber, G.; Liedl, K. R.; Stuppner, H.; Rollinger, J. M. J. Med. Chem. 2010, 53, 778–786. doi:10.1021/jm901440f |
| 5. | Kirchmair, J.; Rollinger, J. M.; Liedl, K. R.; Seidel, N.; Krumbholz, A.; Schmidtke, M. Future Med. Chem. 2011, 3, 437–450. doi:10.4155/fmc.10.292 |
| 19. | Wang, J.-J.; Hu, Z.-P.; Lou, C.-L.; Liu, J.-L.; Li, X.-M.; Yan, M. Tetrahedron 2011, 67, 4578–4583. doi:10.1016/j.tet.2011.04.086 |
| 20. | Zhu, X.; Lin, A.; Shi, Y.; Guo, J.; Zhu, C.; Cheng, Y. Org. Lett. 2011, 13, 4382–4385. doi:10.1021/ol201715h |
| 21. | Li, X.-M.; Wang, B.; Zhang, J.-M.; Yan, M. Org. Lett. 2011, 13, 374–377. doi:10.1021/ol102570b |
| 22. | Xie, J.-W.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.-G.; Chen, Y.-C. Org. Lett. 2007, 9, 413–415. doi:10.1021/ol062718a |
| 23. | Liu, Y.; Liu, X.; Wang, M.; He, P.; Lin, L.; Feng, X. J. Org. Chem. 2012, 77, 4136–4142. doi:10.1021/jo3001047 |
| 3. | Yang, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tai, T.; Nunoura, Y.; Watanabe, K. J. Nat. Prod. 1999, 62, 1672–1674. doi:10.1021/np990096e |
| 22. | Xie, J.-W.; Yue, L.; Chen, W.; Du, W.; Zhu, J.; Deng, J.-G.; Chen, Y.-C. Org. Lett. 2007, 9, 413–415. doi:10.1021/ol062718a |
| 12. | den Hartog, T.; Harutyunyan, S. R.; Font, D.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed. 2008, 47, 398–401. doi:10.1002/anie.200703702 |
| 13. | Bernardi, L.; López-Cantarero, J.; Niess, B.; Jørgensen, K. A. J. Am. Chem. Soc. 2007, 129, 5772–5778. doi:10.1021/ja0707097 |
| 14. | Murphy, J. J.; Quintard, A.; McArdle, P.; Alexakis, A.; Stephens, J. C. Angew. Chem., Int. Ed. 2011, 50, 5095–5098. doi:10.1002/anie.201100804 |
| 15. | Tian, X.; Liu, Y.; Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 6439–6442. doi:10.1002/anie.201202392 |
| 17. | Katritzky, A. R.; Wang, Z.; Wang, M.; Hall, C. D.; Suzuki, K. J. Org. Chem. 2005, 70, 4854–4856. doi:10.1021/jo050307m |
| 10. | Csákÿ, A. G.; de la Herrán, G.; Murcia, M. C. Chem. Soc. Rev. 2010, 39, 4080–4102. doi:10.1039/b924486g |
| 11. | Silva, E. M. P.; Silva, A. M. S. Synthesis 2012, 44, 3109–3128. doi:10.1055/s-0032-1316778 |
| 18. | Baranovsky, A.; Schmitt, B.; Fowler, D. J.; Schneider, B. Synth. Commun. 2003, 33, 1019–1045. doi:10.1081/SCC-120016367 |
| 9. | Kuroyanagi, M.; Noro, T.; Fukushima, S.; Aiyama, R.; Ikuta, A.; Itokawa, H.; Morita, M. Chem. Pharm. Bull. 1983, 31, 1544–1550. doi:10.1248/cpb.31.1544 |
| 32. | The ee value of the synthetic (+)-1and (−)-1 was determined to be 80% and 81%, respectively, suggesting that partial racemization occurred in the HWE reaction, presumably via the intermediate 8. |
| 8. | Yang, C.-S.; Wang, X.-B.; Wang, J.-S.; Luo, J.-G.; Luo, L.; Kong, L.-Y. Org. Lett. 2011, 13, 3380–3383. doi:10.1021/ol201137v |
| 16. | McLeod, M. C.; Wilson, Z. E.; Brimble, M. A. Org. Lett. 2011, 13, 5382–5385. doi:10.1021/ol202265g |
© 2013 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)