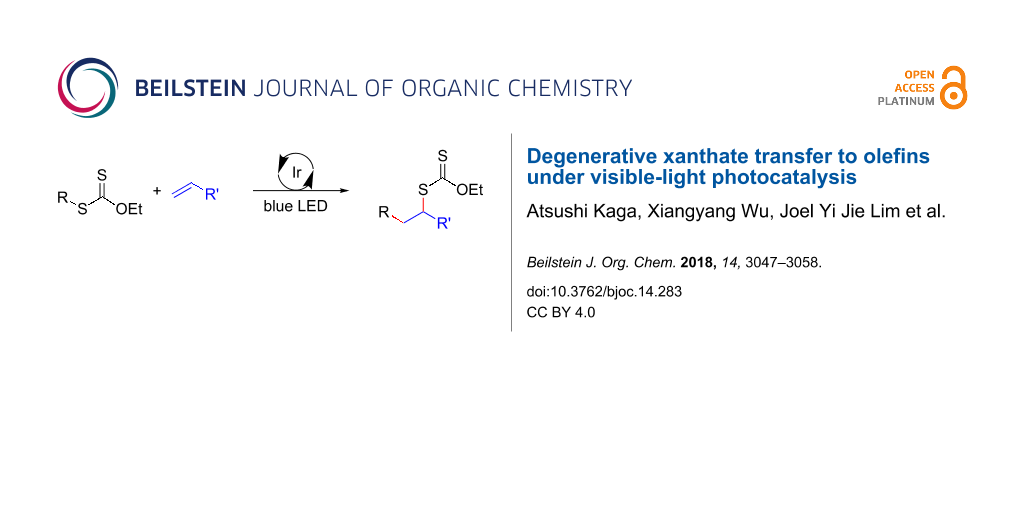
Abstract
The degenerative transfer of xanthates to olefins is enabled by the iridium-based photocatalyst [Ir{dF(CF3)ppy}2(dtbbpy)](PF6) under blue LED light irradiation. Detailed mechanistic investigations through kinetics and photophysical studies revealed that the process operates under a radical chain mechanism, which is initiated through triplet-sensitization of xanthates by the long-lived triplet state of the iridium-based photocatalyst.

Graphical Abstract
Introduction
A degenerative radical transfer of xanthates to olefins has been developed as a robust synthetic tool to create new C–C and C–S bonds in a single operation [1-13]. The method is featured by not only its capability of introducing a wide range of carbon substituents but also the ability of the installed xanthyl group in being transformed into a variety of functionalities [1-14]. This concept has also been of particular importance in the field of polymer science, known as reversible addition–fragmentation chain transfer (RAFT) polymerization [15,16]. Mechanistically, the degenerative transfer of xanthates 1 to olefins 2 proceeds through a radical chain mechanism, and thus requires an initial formation of carbon radicals A that add onto olefins 2. The subsequent reaction of the resulting alkyl radicals B with xanthates 1 provides xanthate adducts 3 with generation of carbon radicals A that maintain the radical chain (Scheme 1A). Peroxide initiators such as dilauroyl peroxide (DLP) are commonly utilized [1-14], while decomposition of DLP needs a high reaction temperature and inevitably generates considerable amounts of byproducts derived from DLP that sometimes require tedious purification of the desired products. A combination of triethylborane (Et3B) and molecular oxygen can also initiate the reaction at lower temperature (e.g., room temperature), while the employment of Et3B is hampered due to its pyrophoric nature under aerobic conditions as well as undesired Et3B-mediated dexanthylation of α-xanthyl ketones [17-21]. As an alternative strategy, a light-driven approach has been developed [22-26], since the first degenerative transfer of xanthates using S-benzoyl O-ethyl xanthate as a photo-cleavable initiator under tungsten lamp irradiation was reported by Zard [25,26] (Scheme 1B). However, these protocols have thus far adopted energy intensive light sources. Therefore, there is still ample room for establishing new protocols to realize the degenerative transfer of xanthates onto olefins under user-friendly and milder reaction conditions. Herein, we report a photocatalytic degenerative radical transfer of xanthates to olefins using an iridium-based photocatalyst under blue LED irradiation (Scheme 1C). A series of mechanistic investigations identified that the process involves a triplet-sensitization of the xanthates by the long-lived triplet state of the iridium-based photocatalyst that triggers the radical chain process [27].
![[1860-5397-14-283-i1]](/bjoc/content/inline/1860-5397-14-283-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Degenerative radical transfer of xanthates to olefins.
Scheme 1: Degenerative radical transfer of xanthates to olefins.
Results and Discussion
Over the last decade, there has been a remarkable advance in synthetic chemistry that takes advantage of various chromophores (either metallic or organic) having visible-light charge transfer absorption [28-37]. In the area of polymer synthesis, visible-light-induced RAFT polymerization of xanthates with vinyl monomers under blue LED (light-emitting diode) irradiation has been reported [38-41]. Visible-light-induced single unit monomer insertion of the thiocarbonylthio compounds has also been developed for the synthesis of the sequence-controlled oligomers [41-45]. For example, the group of Boyer and Xu developed fac-Ir(ppy)3 (6)-catalyzed polymerization of xanthate 4 with various vinyl monomers such as vinyl acetate, providing polymers of type 5 having a high molecular weight with a narrow molecular weight distribution. It was proposed that the polymerization is initiated by single-electron reduction of xanthate 4 by the highly reducing photo-excited state of fac-Ir(ppy)3 (6) [46], although the details were not elucidated (Scheme 2) [39,40].
![[1860-5397-14-283-i2]](/bjoc/content/inline/1860-5397-14-283-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Photocatalytic RAFT polymerization of xanthate 4.
Scheme 2: Photocatalytic RAFT polymerization of xanthate 4.
Based on these backgrounds, we wondered if the degenerative transfer of xanthates onto olefins could be facilitated by visible-light photocatalysis under milder reaction conditions. We therefore commenced our investigation with the reaction of ethyl ethoxycarbonylmethyl xanthate (1a) and 1-octene (2a) using fac-Ir(ppy)3 (6) in DMSO under blue LED irradiation (λmax = 469 nm, Table 1, entry 1). As expected, the desired xanthate transfer was observed, while the process efficiency was not very high, forming 3aa in only 58% yield with incomplete conversion even after stirring for 20 h. Interestingly, we found that the employment of the less reducing Ir catalysts 7 [46] and 8 [47] also worked for the process (Table 1, entries 2 and 3). Especially, the rather oxidizing [Ir{dF(CF3)ppy}2(dtbpy)](PF6) (8) resulted in full conversion of 1a, affording 3aa in 89% yield (Table 1, entry 3). Other photocatalysts, such as Ru(bpy)3Cl2 (9) [46], fluorescein (10) [48], and phenoxazine 11 [49], were not optimal for the present transformation (Table 1, entries 4–6). It should be noted that the reaction without the photocatalyst under visible light- and halogen lamp irradiation resulted in poorer conversion with formation of 3aa in only 10% and 30% yield, respectively, suggesting that the photocatalyst was important for the degenerative transfer of xanthate 1a (Table 1, entries 7 and 8). On the other hand, the employment of a 365 nm UV lamp in place of the blue LED gave 3aa in 75% yield, although a slower reaction rate was observed compared to the optimal reaction conditions (Table 1, entry 9).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-283-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Photocat. |
E1/2(M+/M*)
[V vs SCE]b |
ET
[kcal/mol]b |
Conv.
[%]c |
Yield
[%]c |
| 1 | 6 | –1.73 | 55.2 | 64 | 58 |
| 2 | 7 | –1.28 | 60.1 | 38 | 34 |
| 3 | 8 | –0.89 | 60.1 | >99 | 90 (89)d |
| 4 | 9 | –0.81 | 46.5 | 13 | 13 |
| 5 | 10 | –1.42 | 44.7 | 34 | 31 |
| 6 | 11 | –1.80 | 56.5 | 9 | 9 |
| 7 | none | – | – | 10 | 10 |
| 8e,f | none | – | – | 39 | 30 |
| 9g | none | – | – | 84 | 75 |
aThe reactions were conducted using 0.3–0.5 mmol of xanthate 1a, 1-octene (2a, 2 equiv) and a photocatalyst in DMSO (1 M) at <30 °C with irradiation of a blue LED strip (λmax = 469 nm, 15 W/m, 1.5 m) under an argon atmosphere. bThe values were obtained from references [46-50]. cNMR yields using 1,1,2,2-tetrachloroethane as an internal standard. dIsolated yield is stated in parentheses. eHalogen lamp (300 W) was used in place of blue LED. fReaction was conducted at 40 °C. g365 nm UV lamp (100 W) was used in place of blue LED.
In principle, visible-light-mediated photocatalysis can serve for electron transfer (for either oxidation or reduction) and/or for energy transfer. We found that the reduction potential Ep/2 of xanthate 1a is −1.78 V vs SCE, which is not sufficient to be reduced by the photoexcited states of Ir catalysts 6–8 (E1/2 of 6*, 7* and 8* = −1.73, −1.28, and −0.89 V vs SCE, respectively [46,47]). Apparently, photoinduced single-electron reduction of xanthate 1a by the photoexcited state of the optimal catalyst 8 is not feasible. In contrast, the triplet energy ET of xanthate 1a was estimated as 57.5 kcal/mol by DFT calculation, that is close to those of photocatalysts 7 and 8 (ET = 60.1 kcal/mol [50]), indicating that the process could be initiated by the triplet sensitization pathway [51,52]. This assumption is in agreement with the lower process efficiency (Table 1, entry 1) observed in the reaction with fac-Ir(ppy)3 (6) that possesses a lower triplet energy (ET = 55.2 kcal/mol [50]). The optimal photocatalyst 8 [47] has a longer excited state lifetime than 7 does [46], suggesting that the lifetime of the excited state of the photocatalyst is a key factor for the energy transfer mechanism.
To obtain a detailed mechanistic insight, steady-state photoluminescence (PL) quenching of photocatalyst 8 was examined using xanthate 1a and 1-octene (2a) as potential quenchers (Figure 1). The intensity of the PL peak of photocatalyst 8 (concentration of 8 was fixed as 25 μM solution in degassed DMSO for all the samples; for details see Supporting Information File 1) at 480 nm, arising from the radiative emission of the 3MLCT state of the photocatalyst, was measured using 410 nm light excitation. When the concentration of xanthate 1a was gradually increased, a reduction in the PL intensity (I) of photocatalyst 8 was observed (Figure 1A). The Stern–Volmer plot of the ratio I0/I, where I0 is the initial PL intensity in the absence of quencher, versus concentration of 1a showed a linear relationship with a quenching rate kq = 1.25 × 107 M−1s−1 (see Supporting Information File 1). On the other hand, the addition of 1-octene (2a, 40 mM), in place of xanthate 1a, resulted in only a small PL quenching of photocatalyst 8 (<8%, Figure 1B).
![[1860-5397-14-283-1]](/bjoc/content/figures/1860-5397-14-283-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Photoluminescence (PL) spectra of the 3MLCT state of 8 in degassed DMSO solvent with (A) various concentrations of xanthate 1a added and (B) 40 mM of 1-octene (2a) added. The inset of (A) gives the Stern–Volmer plot of the corrected PL quenching at 480 nm. (C) Time-resolved PL lifetime decay profiles of photocatalyst 8 in degassed DMSO in the absence of quencher (square), presence of xanthate 1a (circle) and presence of 1-octene (2a, triangle). The mono-exponential decay fits are provided.
Figure 1: Photoluminescence (PL) spectra of the 3MLCT state of 8 in degassed DMSO solvent with (A) various co...
The time-resolved PL lifetime decay profiles of photocatalyst 8 (25 μM solution in degassed DMSO, 410 nm pulse excitation and monitoring emission at 480 nm) were recorded in the absence of a quencher, and in the presence of xanthate 1a and 1-octene (2a, 40 mM, Figure 1C). The lifetime profiles were described using a mono-exponential decay function with a lifetime of 1.40 μs in the absence of a quencher, and 1.03 and 1.37 μs in the presence of xanthate 1a and 1-octene (2a), respectively. The decrease in the PL lifetime of photocatalyst 8 in the presence of xanthate 1a suggests that they are interacting with each other. On the other hand, only a very weak interaction exists between 1-octene (2a) and the photocatalyst 8 as demonstrated by the insignificant PL quenching of the photocatalyst [53].
The ns-transient absorption (TA) spectra of photocatalyst 8 (25 μM solution in degassed DMSO) obtained using 355 nm pulse excitation and recorded at different delay times are shown in Figure 2A. The band between 450 nm and 600 nm is attributed to the excited 3MLCT state of the photocatalyst [53-55]. The positive ΔOD feature in the UV region (<400 nm) is also ascribed to the excited 3MLCT state [55]. The transient kinetic profile probed at 480 nm decays mono-exponentially with a lifetime of 1.73 μs (Figure 2B); close to the lifetime of the excited 3MLCT state of photocatalyst 8 measured in Figure 1C. The ns-TA spectra of xanthate 1a in degassed DMSO, measured using 355 nm pulse excitation, at various delay times show a broad band centered at ≈620 nm (Figure 2C). This band has previously been ascribed to the absorption of the xanthic acid radical formed plausibly from homolytic C–S bond cleavage of the short-lived triplet state of xanthate 1a [22].
![[1860-5397-14-283-2]](/bjoc/content/figures/1860-5397-14-283-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (A) ns-Transient absorption spectra of photocatalyst 8 in degassed DMSO recorded at different delay times (excitation wavelength = 355 nm). (B) ns-TA kinetic profile probed at 480 nm for photocatalyst 8 in the absence and presence of xanthate 1a. (C) and (D) ns-Transient absorption spectra of xanthate 1a and 8 in the presence of 1a in degassed DMSO recorded at different delay times, respectively (excitation wavelength = 355 nm).
Figure 2: (A) ns-Transient absorption spectra of photocatalyst 8 in degassed DMSO recorded at different delay...
The ns-TA spectra of photocatalyst 8 (25 μM) in the presence of xanthate 1a (40 mM) in degassed DMSO at various delay times are shown in Figure 2D. The kinetic profile at 480 nm is described using a mono-exponential decay function with a quenched lifetime of 1.27 μs. The ca. 37% decrease in the lifetime of the excited 3MLCT state of photocatalyst 8 observed in the PL lifetime decay measurement (Figure 1C) and ns-TA kinetic measurement (Figure 2B) can be rationalized using either an energy-transfer or an electron-transfer mechanism. For the same delay times and in the presence of 1a, the ΔOD values in Figure 2D are smaller than those of photocatalyst 8 in the absence of the xanthate (Figure 2A). This is due to the quenching of the 3MLCT state of the photocatalyst. If an electron-transfer process occurs from photocatalyst 8 to xanthate 1a, the ΔOD values in the UV region should be noticeably higher due to the TA band contribution from bpy− connected to an Ir metal center of the +4 oxidation state (i.e., absorption due to [IrIV{dF(CF3)ppy}2](dtbpy−)] species) [55]. However, this was not observed in Figure 2D; suggesting that quenching is due to energy transfer rather than electron transfer, and in agreement with the thermodynamic consideration where single-electron reduction of xanthate 1a likely does not proceed with the excited photocatalyst 8. We therefore propose that the observed PL quenching is due to energy transfer from the excited 3MLCT state of photocatalyst 8 to the triplet state of xanthate 1a. When comparing the normalized TA spectra of photocatalyst 8 in the absence and presence of xanthate 1a (see Supporting Information File 1, Figure S4), an additional contribution from a broad ΔOD band that stretches from 500 nm to 800 nm is seen for the latter which is attributed to the absorption of the xanthic acid radical. In this case, the xanthic acid radical is formed from the homolytic bond cleavage of the excited triplet state of 1a formed by direct 355 nm laser light excitation and triplet–triplet energy transfer involving the excited photocatalyst 8.
To confirm the possibility of a direct photoexcitation of xanthate 1 using blue LED light irradiation as an alternative mechanism, a steady-state UV–vis absorption spectroscopy study of xanthate 1a was conducted (Figure 3). The UV–vis absorption spectrum of 1a (1 mM in DMSO) showed absorption bands at 340–390 nm assigned to the n→π* electronic transition of the C=S bond as a characteristic peak of thiocarbonyl containing compounds [56]. In fact, the reaction of 1a and 2a under 365 nm UV lamp irradiation without a photocatalyst delivered product 3aa in 75% yield (Table 1, entry 9). This indicates the excitation of xanthate 1a through an n→π* electronic transition of the C=S bond is in operation in the UV irradiation process.
![[1860-5397-14-283-3]](/bjoc/content/figures/1860-5397-14-283-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis absorption spectrum of 1a (1 mM solution in DMSO).
Figure 3: UV–vis absorption spectrum of 1a (1 mM solution in DMSO).
The involvement of a radical chain mechanism was further confirmed by calculating the quantum yield (Φ) because a chain process provides multiple equivalents of product per photon absorbed by the photocatalyst (Φ > 1). The photon flux of blue LED (λmax = 469 nm) was determined using the potassium ferrioxalate actinometer [57,58]. After irradiation of the mixture of xanthate 1a and olefin 2b under optimal reaction conditions with blue LED light irradiation for 4 h (Scheme 3), product 3ab was obtained in 58% yield. This is consistent with 12 equivalents of xanthate adduct 3ab produced per photon absorbed by the photocatalyst 8 (Φ = 12).
![[1860-5397-14-283-i3]](/bjoc/content/inline/1860-5397-14-283-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Determination of quantum yield.
Scheme 3: Determination of quantum yield.
On the basis of these observations, a proposed triplet sensitization mechanism is illustrated in Scheme 4. In this reaction, photocatalyst 8 serves as a catalyst of an initiation step through energy transfer from photoexcited 8* to xanthate 1 to form excited xanthate 1* and regeneration of 8 in the ground state [59-61]. The resulting excited xanthate 1* induces homolytic scission of the C–S bond to generate the stabilized S-radical and C-radical A, which then enters the innate radical chain-propagation mechanism to provide xanthate adduct 3. It is worth noting that at the wavelength of the light source used (469 nm), xanthate 1a absorbs a negligible amount of light (Figure 3) and the majority of triplet 1a formed is due to energy transfer from excited catalyst 8*.
![[1860-5397-14-283-i4]](/bjoc/content/inline/1860-5397-14-283-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Having optimized the reaction conditions on the photocatalytic degenerative transfer of xanthates, we next explored the scope of olefins 2 using xanthate 1a (Table 2). The present method tolerated a variety of functionalities such as acetyl, cyano, silyl, ethoxy, N-Boc amino, boryl, hydroxy, and halogen groups, affording xanthate adducts 3ab–ai in good yields (Table 2, entries 1–8). We found strained 1,1-disubstituted olefins such as methylenecyclopropane 2j and methylenecyclobutane (2k) are amenable to the current protocol (Table 2, entries 9 and 10), whereas the reaction of methylenecyclopentane (2l) afforded not only the desired xanthate adduct 3al in 60% yield but also the substituted cyclopentene 3al’ in 18% yield (Table 2, entry 11), implicating that the redox process is partially operating along with the main radical chain process. Norbornene (2m) was found reactive for degenerative transfer of xanthate 1a (Table 2, entry 12). The reaction was also applied to dienes 2n and 2o, which led the formation of functionalized cyclopentane 3an and pyrrolidine 3ao, respectively, via 5-exo-trig radical cyclization (Table 2, entries 13 and 14). The present method was capable in functionalizing olefins 2p and 2q installed on steroid scaffolds with high efficiency (Table 2, entries 15 and 16).
Table 2: Scope of olefins.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-283-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | Olefins 2 | Products 3 | Yieldb (time) | |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-283-i7.svg?max-width=637&scale=1.0)
2 |
R |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-283-i8.svg?max-width=637&scale=1.0)
|
||
| 1 | 2b | OAc | 3ab | 88% (18 h) |
| 2 | 2c | CN | 3ac | 73% (45 h) |
| 3 | 2d | SiMe3 | 3ad | 85% (46 h) |
| 4c | 2e | OEt | 3ae | 70% (41 h) |
| 5c | 2f | NHBoc | 3af | 80% (61 h) |
| 6d | 2g | Bpin | 3ag | 72% (30 h) |
| 7 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-283-i9.svg?max-width=637&scale=1.0)
2h |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-283-i10.svg?max-width=637&scale=1.0)
3ah |
84% (37 h) | |
| 8 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-283-i11.svg?max-width=637&scale=1.0)
2i |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-283-i12.svg?max-width=637&scale=1.0)
3ai |
73% (48 h) | |
| 9 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-283-i13.svg?max-width=637&scale=1.0)
2j |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-283-i14.svg?max-width=637&scale=1.0)
3aj |
64% (48 h) | |
| 10 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-283-i15.svg?max-width=637&scale=1.0)
2k |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-283-i16.svg?max-width=637&scale=1.0)
3ak |
90% (26 h) | |
| 11d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-283-i17.svg?max-width=637&scale=1.0)
2l |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-283-i18.svg?max-width=637&scale=1.0)
3al |
60% (20 h) | |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-283-i19.svg?max-width=637&scale=1.0)
3al’ |
18%e (20 h)
endo/exo = 80:20 |
|||
| 12 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-283-i20.svg?max-width=637&scale=1.0)
2m |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-283-i21.svg?max-width=637&scale=1.0)
3am |
73% (10 h)
(dr = 78:22) |
|
| 13 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-283-i22.svg?max-width=637&scale=1.0)
2n |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-283-i23.svg?max-width=637&scale=1.0)
3an |
68% (24 h)
(dr = 88:12) |
|
| 14 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-283-i24.svg?max-width=637&scale=1.0)
2o |
![[Graphic 21]](/bjoc/content/inline/1860-5397-14-283-i25.svg?max-width=637&scale=1.0)
3ao |
74% (13 h)
(dr = 71:29) |
|
| 15f |
![[Graphic 22]](/bjoc/content/inline/1860-5397-14-283-i26.svg?max-width=637&scale=1.0)
2p |
![[Graphic 23]](/bjoc/content/inline/1860-5397-14-283-i27.svg?max-width=637&scale=1.0)
3ap |
94% (26 h)
(dr = 50:50) |
|
| 16g |
![[Graphic 24]](/bjoc/content/inline/1860-5397-14-283-i28.svg?max-width=637&scale=1.0)
2q |
![[Graphic 25]](/bjoc/content/inline/1860-5397-14-283-i29.svg?max-width=637&scale=1.0)
3aq |
80% (50 h)
(dr = 50:50) |
|
aThe reactions were conducted using xanthate 1a (0.3–0.5 mmol), olefin 2 (2 equiv) and 8 (0.5 mol %) in DMSO (1 M) at <30 °C with irradiation of a blue LED strip (λmax = 469 nm) under an argon atmosphere. bIsolated yields are stated. c1 mol % of 8 was used. d4 equiv of olefin 2 were used. eNMR yield using 1,1,2,2-tetrachloroethane as an internal standard. fThe reaction was conducted in DMSO/DCE 1:1 (0.5 M). gThe reaction was conducted in DMSO/DCE 3:5 (0.4 M).
We next examined the reactions of various xanthates 1 with allyl acetate (2b, Table 3). The reactions of ketonyl xanthates having phenyl, para-bromophenyl, methyl, cyclopropyl, N,O-dimethyl acetylhydroxamate, and chloromethyl moieties proceeded smoothly, producing xanthate adducts 3bb–gb in good yields (Table 3, entries 1–6). Notably, the photocatalytically cleavable aryl bromide (Table 3, entry 2) and the α-chlorocarbonyl moiety (Table 3, entry 6) were also stable under the current reaction conditions [62,63]. Furthermore, double addition of bisxanthate 1h was successfully achieved in the presence of 5 equiv of olefin 2b, giving 3hb in 69% yield (Table 3, entry 7). This method is also suitable for generating α-aminoalkyl radicals from phthalimidomethyl and succinimidomethyl xanthates [64], as well as α-trifluoromethylamino xanthate 1k [65] to afford desired products 3ib–kb in good to moderate yields (Table 3, entries 8–10).
Table 3: Scope of xanthates.a
![[Graphic 26]](/bjoc/content/inline/1860-5397-14-283-i30.svg?max-width=637&scale=1.0)
|
|||
| Entry | Xanthates 1 | Products 3 | Yieldb (time) |
| 1 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-14-283-i31.svg?max-width=637&scale=1.0)
1b |
![[Graphic 28]](/bjoc/content/inline/1860-5397-14-283-i32.svg?max-width=637&scale=1.0)
3bb |
78% (44 h) |
| 2 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-14-283-i33.svg?max-width=637&scale=1.0)
1c |
![[Graphic 30]](/bjoc/content/inline/1860-5397-14-283-i34.svg?max-width=637&scale=1.0)
3cb |
69% (47 h) |
| 3 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-14-283-i35.svg?max-width=637&scale=1.0)
1d |
![[Graphic 32]](/bjoc/content/inline/1860-5397-14-283-i36.svg?max-width=637&scale=1.0)
3db |
59% (51 h) |
| 4c |
![[Graphic 33]](/bjoc/content/inline/1860-5397-14-283-i37.svg?max-width=637&scale=1.0)
1e |
![[Graphic 34]](/bjoc/content/inline/1860-5397-14-283-i38.svg?max-width=637&scale=1.0)
3eb |
82% (71 h) |
| 5 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-14-283-i39.svg?max-width=637&scale=1.0)
1f |
![[Graphic 36]](/bjoc/content/inline/1860-5397-14-283-i40.svg?max-width=637&scale=1.0)
3fb |
81% (27 h) |
| 6 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-14-283-i41.svg?max-width=637&scale=1.0)
1g |
![[Graphic 38]](/bjoc/content/inline/1860-5397-14-283-i42.svg?max-width=637&scale=1.0)
3gb |
75% (17 h) |
| 7d |
![[Graphic 39]](/bjoc/content/inline/1860-5397-14-283-i43.svg?max-width=637&scale=1.0)
1h |
![[Graphic 40]](/bjoc/content/inline/1860-5397-14-283-i44.svg?max-width=637&scale=1.0)
3hb |
69% (52 h)
(dr = 52:48) |
| 8 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-14-283-i45.svg?max-width=637&scale=1.0)
1i |
![[Graphic 42]](/bjoc/content/inline/1860-5397-14-283-i46.svg?max-width=637&scale=1.0)
3ib |
80% (41 h) |
| 9 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-14-283-i47.svg?max-width=637&scale=1.0)
1j |
![[Graphic 44]](/bjoc/content/inline/1860-5397-14-283-i48.svg?max-width=637&scale=1.0)
3jb |
56% (24 h) |
| 10 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-14-283-i49.svg?max-width=637&scale=1.0)
1k |
![[Graphic 46]](/bjoc/content/inline/1860-5397-14-283-i50.svg?max-width=637&scale=1.0)
3kb |
74% (24 h)
(dr = 63:37) |
aThe reactions were conducted using xanthate 1 (0.3 mmol), olefin 2b (2 equiv) and 8 (0.5 mol %) in DMSO (1 M) at <30 °C with irradiation of a blue LED strip (λmax = 469 nm) under an argon atmosphere. bIsolated yields are stated. c1 mol % of 8 was used. dFive equivalents of olefin 2b were used.
Conclusion
We have established a protocol for a photoinduced radical addition of xanthates to olefins using an iridium-based photocatalyst under blue LED irradiation, leading to diverse xanthate adducts. This reaction proceeds through a radical-chain propagation mechanism via an initiation involving a triplet-sensitization process of xanthates by an excited iridium-based photocatalyst.
Supporting Information
| Supporting Information File 1: Full experimental details and analytical data. | ||
| Format: PDF | Size: 4.9 MB | Download |
References
-
Zard, S. Z. Acc. Chem. Res. 2018, 51, 1722–1733. doi:10.1021/acs.accounts.8b00201
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Synlett 2017, 28, 2685–2696. doi:10.1055/s-0036-1590809
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Isr. J. Chem. 2017, 57, 202–217. doi:10.1002/ijch.201600094
Return to citation in text: [1] [2] [3] -
Zard, S. Z. Org. Biomol. Chem. 2016, 14, 6891–6912. doi:10.1039/c6ob01087c
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Synlett 2016, 27, 680–701. doi:10.1055/s-0035-1561300
Return to citation in text: [1] [2] [3] -
Debien, L.; Quiclet-Sire, B.; Zard, S. Z. Acc. Chem. Res. 2015, 48, 1237–1253. doi:10.1021/acs.accounts.5b00019
Return to citation in text: [1] [2] [3] -
Zard, S. Z. J. Phys. Org. Chem. 2012, 25, 953–964. doi:10.1002/poc.2976
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Chimia 2012, 66, 404–412. doi:10.2533/chimia.2012.404
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Pure Appl. Chem. 2011, 83, 519–551. doi:10.1351/pac-con-10-08-07
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Top. Curr. Chem. 2006, 264, 201–236. doi:10.1007/128_029
Return to citation in text: [1] [2] [3] -
Quiclet-Sire, B.; Zard, S. Z. Chem. – Eur. J. 2006, 12, 6002–6016. doi:10.1002/chem.200600510
Return to citation in text: [1] [2] [3] -
Zard, S. Z. Aust. J. Chem. 2006, 59, 663–668. doi:10.1071/ch06263
Return to citation in text: [1] [2] [3] -
Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721
Return to citation in text: [1] [2] [3] -
Czaplyski, W. L.; Na, C. G.; Alexanian, E. J. J. Am. Chem. Soc. 2016, 138, 13854–13857. doi:10.1021/jacs.6b09414
Return to citation in text: [1] [2] -
Perrier, S. Macromolecules 2017, 50, 7433–7447. doi:10.1021/acs.macromol.7b00767
Return to citation in text: [1] -
Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, 5559–5562. doi:10.1021/ma9804951
Return to citation in text: [1] -
García-Merinos, J. P.; Hernández-Pérez, J. P.; Martínez-García, L.; Rojas-Lima, S.; López-Ruiz, H. J. Mex. Chem. Soc. 2007, 51, 209–212.
Return to citation in text: [1] -
Boivin, J.; Nguyen, V. T. Beilstein J. Org. Chem. 2007, 3, No. 45. doi:10.1186/1860-5397-3-45
Return to citation in text: [1] -
Charrier, N.; Gravestock, D.; Zard, S. Z. Angew. Chem., Int. Ed. 2006, 45, 6520–6523. doi:10.1002/anie.200601567
Return to citation in text: [1] -
Jean-Baptiste, L.; Yemets, S.; Legay, R.; Lequeux, T. J. Org. Chem. 2006, 71, 2352–2359. doi:10.1021/jo052528y
Return to citation in text: [1] -
Briggs, M. E.; Zard, S. Z. Synlett 2005, 334–336. doi:10.1055/s-2004-837191
Return to citation in text: [1] -
Tazhe Veetil, A.; Šolomek, T.; Ngoy, B. P.; Pavlíková, N.; Heger, D.; Klán, P. J. Org. Chem. 2011, 76, 8232–8242. doi:10.1021/jo201385b
Return to citation in text: [1] [2] -
Ferjančić, Z.; Čeković, Ž.; Saičić, R. N. Tetrahedron Lett. 2000, 41, 2979–2982. doi:10.1016/s0040-4039(00)00286-0
Return to citation in text: [1] -
Maslak, V.; Čeković, Ž.; Saičić, R. N. Synlett 1998, 1435–1437. doi:10.1055/s-1998-1946
Return to citation in text: [1] -
Mestre, F.; Tailham, C.; Zard, S. Z. Heterocycles 1989, 28, 171–174. doi:10.3987/com-88-s77
Return to citation in text: [1] [2] -
Delduc, P.; Tailhan, C.; Zard, S. Z. J. Chem. Soc., Chem. Commun. 1988, 308–310. doi:10.1039/c39880000308
Return to citation in text: [1] [2] -
López-Mendoza, P.; Díaz, J. E.; Loaiza, A. E.; Miranda, L. D. Tetrahedron 2018, 74, 5494–5502. doi:10.1016/j.tet.2018.04.079
Return to citation in text: [1] -
Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, No. 0052. doi:10.1038/s41570-017-0052
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057
Return to citation in text: [1] -
Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035–10074. doi:10.1021/acs.chemrev.6b00018
Return to citation in text: [1] -
Ravelli, D.; Protti, S.; Fagnoni, M. Chem. Rev. 2016, 116, 9850–9913. doi:10.1021/acs.chemrev.5b00662
Return to citation in text: [1] -
Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760
Return to citation in text: [1] -
Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449
Return to citation in text: [1] -
Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Chem. Soc. Rev. 2016, 45, 6165–6212. doi:10.1039/c6cs00185h
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n
Return to citation in text: [1] -
Ding, C.; Fan, C.; Jiang, G.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Macromol. Rapid Commun. 2015, 36, 2181–2185. doi:10.1002/marc.201500427
Return to citation in text: [1] -
Shanmugam, S.; Xu, J.; Boyer, C. Macromolecules 2014, 47, 4930–4942. doi:10.1021/ma500842u
Return to citation in text: [1] [2] -
Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. J. Am. Chem. Soc. 2014, 136, 5508–5519. doi:10.1021/ja501745g
Return to citation in text: [1] [2] -
Phommalysack-Lovan, J.; Chu, Y.; Boyer, C.; Xu, J. Chem. Commun. 2018, 54, 6591–6606. doi:10.1039/c8cc02783h
Return to citation in text: [1] [2] -
Huang, Z.; Noble, B. B.; Corrigan, N.; Chu, Y.; Satoh, K.; Thomas, D. S.; Hawker, C. J.; Moad, G.; Kamigaito, M.; Coote, M. L.; Boyer, C.; Xu, J. J. Am. Chem. Soc. 2018, 140, 13392–13406. doi:10.1021/jacs.8b08386
Return to citation in text: [1] -
Aerts, A.; Lewis, R. W.; Zhou, Y.; Malic, N.; Moad, G.; Postma, A. Macromol. Rapid Commun. 2018, 39, 1800240. doi:10.1002/marc.201800240
Return to citation in text: [1] -
Fu, C.; Huang, Z.; Hawker, C. J.; Moad, G.; Xu, J.; Boyer, C. Polym. Chem. 2017, 8, 4637–4643. doi:10.1039/c7py00713b
Return to citation in text: [1] -
Xu, J.; Fu, C.; Shanmugam, S.; Hawker, C. J.; Moad, G.; Boyer, C. Angew. Chem., Int. Ed. 2017, 56, 8376–8383. doi:10.1002/anie.201610223
Return to citation in text: [1] -
Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+
Return to citation in text: [1] [2] [3] [4] -
Shen, T.; Zhao, Z.-G.; Yu, Q.; Xu, H.-J. J. Photochem. Photobiol., A 1989, 47, 203–212. doi:10.1016/1010-6030(89)87066-2
Return to citation in text: [1] [2] -
Du, Y.; Pearson, R. M.; Lim, C.-H.; Sartor, S. M.; Ryan, M. D.; Yang, H.; Damrauer, N. H.; Miyake, G. M. Chem. – Eur. J. 2017, 23, 10962–10968. doi:10.1002/chem.201702926
Return to citation in text: [1] [2] -
Singh, A.; Teegardin, K.; Kelly, M.; Prasad, K. S.; Krishnan, S.; Weaver, J. D. J. Organomet. Chem. 2015, 776, 51–59. doi:10.1016/j.jorganchem.2014.10.037
Return to citation in text: [1] [2] [3] -
Strieth-Kalthoff, F.; James, M. J.; Teders, M.; Pitzer, L.; Glorius, F. Chem. Soc. Rev. 2018, 47, 7190–7202. doi:10.1039/c8cs00054a
Return to citation in text: [1] -
Xiao, W.-J.; Zhou, Q.-Q.; Zou, Y.-Q.; Lu, L.-Q. Angew. Chem., Int. Ed. 2018. doi:10.1002/anie.201803102
Return to citation in text: [1] -
Teders, M.; Henkel, C.; Anhäuser, L.; Strieth-Kalthoff, F.; Gómez-Suárez, A.; Kleinmans, R.; Kahnt, A.; Rentmeister, A.; Guldi, D.; Glorius, F. Nat. Chem. 2018, 10, 981–988. doi:10.1038/s41557-018-0102-z
Return to citation in text: [1] [2] -
van As, D. J.; Connell, T. U.; Brzozowski, M.; Scully, A. D.; Polyzos, A. Org. Lett. 2018, 20, 905–908. doi:10.1021/acs.orglett.7b03565
Return to citation in text: [1] -
Ichimura, K.; Kobayashi, T.; King, K. A.; Watts, R. J. J. Phys. Chem. 1987, 91, 6104–6106. doi:10.1021/j100308a012
Return to citation in text: [1] [2] [3] -
Coyle, J. D. Tetrahedron 1985, 41, 5393–5425. doi:10.1016/s0040-4020(01)91341-9
Return to citation in text: [1] -
Cismesia, M. A.; Yoon, T. P. Chem. Sci. 2015, 6, 5426–5434. doi:10.1039/c5sc02185e
Return to citation in text: [1] -
Hatchard, C. G.; Parker, C. A. Proc. R. Soc. London, Ser. A 1956, 235, 518–536. doi:10.1098/rspa.1956.0102
Return to citation in text: [1] -
Studer, A.; Curran, D. P. Angew. Chem., Int. Ed. 2016, 55, 58–102. doi:10.1002/anie.201505090
Return to citation in text: [1] -
Christmann, J.; Ibrahim, A.; Charlot, V.; Croutxé-Barghorn, C.; Ley, C.; Allonas, X. ChemPhysChem 2016, 17, 2309–2314. doi:10.1002/cphc.201600034
Return to citation in text: [1] -
Arceo, E.; Montroni, E.; Melchiorre, P. Angew. Chem., Int. Ed. 2014, 53, 12064–12068. doi:10.1002/anie.201406450
Return to citation in text: [1] -
Devery, J. J., III; Nguyen, J. D.; Dai, C.; Stephenson, C. R. J. ACS Catal. 2016, 6, 5962–5967. doi:10.1021/acscatal.6b01914
Return to citation in text: [1] -
Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599
Return to citation in text: [1] -
Quiclet-Sire, B.; Zard, S. Z. Org. Lett. 2008, 10, 3279–3282. doi:10.1021/ol801162m
Return to citation in text: [1] -
Gagosz, F.; Zard, S. Z. Org. Lett. 2003, 5, 2655–2657. doi:10.1021/ol034812m
Return to citation in text: [1]
| 55. | Ichimura, K.; Kobayashi, T.; King, K. A.; Watts, R. J. J. Phys. Chem. 1987, 91, 6104–6106. doi:10.1021/j100308a012 |
| 22. | Tazhe Veetil, A.; Šolomek, T.; Ngoy, B. P.; Pavlíková, N.; Heger, D.; Klán, P. J. Org. Chem. 2011, 76, 8232–8242. doi:10.1021/jo201385b |
| 55. | Ichimura, K.; Kobayashi, T.; King, K. A.; Watts, R. J. J. Phys. Chem. 1987, 91, 6104–6106. doi:10.1021/j100308a012 |
| 1. | Zard, S. Z. Acc. Chem. Res. 2018, 51, 1722–1733. doi:10.1021/acs.accounts.8b00201 |
| 2. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2017, 28, 2685–2696. doi:10.1055/s-0036-1590809 |
| 3. | Quiclet-Sire, B.; Zard, S. Z. Isr. J. Chem. 2017, 57, 202–217. doi:10.1002/ijch.201600094 |
| 4. | Zard, S. Z. Org. Biomol. Chem. 2016, 14, 6891–6912. doi:10.1039/c6ob01087c |
| 5. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2016, 27, 680–701. doi:10.1055/s-0035-1561300 |
| 6. | Debien, L.; Quiclet-Sire, B.; Zard, S. Z. Acc. Chem. Res. 2015, 48, 1237–1253. doi:10.1021/acs.accounts.5b00019 |
| 7. | Zard, S. Z. J. Phys. Org. Chem. 2012, 25, 953–964. doi:10.1002/poc.2976 |
| 8. | Quiclet-Sire, B.; Zard, S. Z. Chimia 2012, 66, 404–412. doi:10.2533/chimia.2012.404 |
| 9. | Quiclet-Sire, B.; Zard, S. Z. Pure Appl. Chem. 2011, 83, 519–551. doi:10.1351/pac-con-10-08-07 |
| 10. | Quiclet-Sire, B.; Zard, S. Z. Top. Curr. Chem. 2006, 264, 201–236. doi:10.1007/128_029 |
| 11. | Quiclet-Sire, B.; Zard, S. Z. Chem. – Eur. J. 2006, 12, 6002–6016. doi:10.1002/chem.200600510 |
| 12. | Zard, S. Z. Aust. J. Chem. 2006, 59, 663–668. doi:10.1071/ch06263 |
| 13. | Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721 |
| 17. | García-Merinos, J. P.; Hernández-Pérez, J. P.; Martínez-García, L.; Rojas-Lima, S.; López-Ruiz, H. J. Mex. Chem. Soc. 2007, 51, 209–212. |
| 18. | Boivin, J.; Nguyen, V. T. Beilstein J. Org. Chem. 2007, 3, No. 45. doi:10.1186/1860-5397-3-45 |
| 19. | Charrier, N.; Gravestock, D.; Zard, S. Z. Angew. Chem., Int. Ed. 2006, 45, 6520–6523. doi:10.1002/anie.200601567 |
| 20. | Jean-Baptiste, L.; Yemets, S.; Legay, R.; Lequeux, T. J. Org. Chem. 2006, 71, 2352–2359. doi:10.1021/jo052528y |
| 21. | Briggs, M. E.; Zard, S. Z. Synlett 2005, 334–336. doi:10.1055/s-2004-837191 |
| 47. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 1. | Zard, S. Z. Acc. Chem. Res. 2018, 51, 1722–1733. doi:10.1021/acs.accounts.8b00201 |
| 2. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2017, 28, 2685–2696. doi:10.1055/s-0036-1590809 |
| 3. | Quiclet-Sire, B.; Zard, S. Z. Isr. J. Chem. 2017, 57, 202–217. doi:10.1002/ijch.201600094 |
| 4. | Zard, S. Z. Org. Biomol. Chem. 2016, 14, 6891–6912. doi:10.1039/c6ob01087c |
| 5. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2016, 27, 680–701. doi:10.1055/s-0035-1561300 |
| 6. | Debien, L.; Quiclet-Sire, B.; Zard, S. Z. Acc. Chem. Res. 2015, 48, 1237–1253. doi:10.1021/acs.accounts.5b00019 |
| 7. | Zard, S. Z. J. Phys. Org. Chem. 2012, 25, 953–964. doi:10.1002/poc.2976 |
| 8. | Quiclet-Sire, B.; Zard, S. Z. Chimia 2012, 66, 404–412. doi:10.2533/chimia.2012.404 |
| 9. | Quiclet-Sire, B.; Zard, S. Z. Pure Appl. Chem. 2011, 83, 519–551. doi:10.1351/pac-con-10-08-07 |
| 10. | Quiclet-Sire, B.; Zard, S. Z. Top. Curr. Chem. 2006, 264, 201–236. doi:10.1007/128_029 |
| 11. | Quiclet-Sire, B.; Zard, S. Z. Chem. – Eur. J. 2006, 12, 6002–6016. doi:10.1002/chem.200600510 |
| 12. | Zard, S. Z. Aust. J. Chem. 2006, 59, 663–668. doi:10.1071/ch06263 |
| 13. | Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721 |
| 14. | Czaplyski, W. L.; Na, C. G.; Alexanian, E. J. J. Am. Chem. Soc. 2016, 138, 13854–13857. doi:10.1021/jacs.6b09414 |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 15. | Perrier, S. Macromolecules 2017, 50, 7433–7447. doi:10.1021/acs.macromol.7b00767 |
| 16. | Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, 5559–5562. doi:10.1021/ma9804951 |
| 39. | Shanmugam, S.; Xu, J.; Boyer, C. Macromolecules 2014, 47, 4930–4942. doi:10.1021/ma500842u |
| 40. | Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. J. Am. Chem. Soc. 2014, 136, 5508–5519. doi:10.1021/ja501745g |
| 64. | Quiclet-Sire, B.; Zard, S. Z. Org. Lett. 2008, 10, 3279–3282. doi:10.1021/ol801162m |
| 1. | Zard, S. Z. Acc. Chem. Res. 2018, 51, 1722–1733. doi:10.1021/acs.accounts.8b00201 |
| 2. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2017, 28, 2685–2696. doi:10.1055/s-0036-1590809 |
| 3. | Quiclet-Sire, B.; Zard, S. Z. Isr. J. Chem. 2017, 57, 202–217. doi:10.1002/ijch.201600094 |
| 4. | Zard, S. Z. Org. Biomol. Chem. 2016, 14, 6891–6912. doi:10.1039/c6ob01087c |
| 5. | Quiclet-Sire, B.; Zard, S. Z. Synlett 2016, 27, 680–701. doi:10.1055/s-0035-1561300 |
| 6. | Debien, L.; Quiclet-Sire, B.; Zard, S. Z. Acc. Chem. Res. 2015, 48, 1237–1253. doi:10.1021/acs.accounts.5b00019 |
| 7. | Zard, S. Z. J. Phys. Org. Chem. 2012, 25, 953–964. doi:10.1002/poc.2976 |
| 8. | Quiclet-Sire, B.; Zard, S. Z. Chimia 2012, 66, 404–412. doi:10.2533/chimia.2012.404 |
| 9. | Quiclet-Sire, B.; Zard, S. Z. Pure Appl. Chem. 2011, 83, 519–551. doi:10.1351/pac-con-10-08-07 |
| 10. | Quiclet-Sire, B.; Zard, S. Z. Top. Curr. Chem. 2006, 264, 201–236. doi:10.1007/128_029 |
| 11. | Quiclet-Sire, B.; Zard, S. Z. Chem. – Eur. J. 2006, 12, 6002–6016. doi:10.1002/chem.200600510 |
| 12. | Zard, S. Z. Aust. J. Chem. 2006, 59, 663–668. doi:10.1071/ch06263 |
| 13. | Zard, S. Z. Angew. Chem., Int. Ed. Engl. 1997, 36, 672–685. doi:10.1002/anie.199706721 |
| 14. | Czaplyski, W. L.; Na, C. G.; Alexanian, E. J. J. Am. Chem. Soc. 2016, 138, 13854–13857. doi:10.1021/jacs.6b09414 |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 28. | Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, No. 0052. doi:10.1038/s41570-017-0052 |
| 29. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 30. | Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057 |
| 31. | Skubi, K. L.; Blum, T. R.; Yoon, T. P. Chem. Rev. 2016, 116, 10035–10074. doi:10.1021/acs.chemrev.6b00018 |
| 32. | Ravelli, D.; Protti, S.; Fagnoni, M. Chem. Rev. 2016, 116, 9850–9913. doi:10.1021/acs.chemrev.5b00662 |
| 33. | Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760 |
| 34. | Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898–6926. doi:10.1021/acs.joc.6b01449 |
| 35. | Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Chem. Soc. Rev. 2016, 45, 6165–6212. doi:10.1039/c6cs00185h |
| 36. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 37. | Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. doi:10.1039/b913880n |
| 41. | Phommalysack-Lovan, J.; Chu, Y.; Boyer, C.; Xu, J. Chem. Commun. 2018, 54, 6591–6606. doi:10.1039/c8cc02783h |
| 42. | Huang, Z.; Noble, B. B.; Corrigan, N.; Chu, Y.; Satoh, K.; Thomas, D. S.; Hawker, C. J.; Moad, G.; Kamigaito, M.; Coote, M. L.; Boyer, C.; Xu, J. J. Am. Chem. Soc. 2018, 140, 13392–13406. doi:10.1021/jacs.8b08386 |
| 43. | Aerts, A.; Lewis, R. W.; Zhou, Y.; Malic, N.; Moad, G.; Postma, A. Macromol. Rapid Commun. 2018, 39, 1800240. doi:10.1002/marc.201800240 |
| 44. | Fu, C.; Huang, Z.; Hawker, C. J.; Moad, G.; Xu, J.; Boyer, C. Polym. Chem. 2017, 8, 4637–4643. doi:10.1039/c7py00713b |
| 45. | Xu, J.; Fu, C.; Shanmugam, S.; Hawker, C. J.; Moad, G.; Boyer, C. Angew. Chem., Int. Ed. 2017, 56, 8376–8383. doi:10.1002/anie.201610223 |
| 59. | Studer, A.; Curran, D. P. Angew. Chem., Int. Ed. 2016, 55, 58–102. doi:10.1002/anie.201505090 |
| 60. | Christmann, J.; Ibrahim, A.; Charlot, V.; Croutxé-Barghorn, C.; Ley, C.; Allonas, X. ChemPhysChem 2016, 17, 2309–2314. doi:10.1002/cphc.201600034 |
| 61. | Arceo, E.; Montroni, E.; Melchiorre, P. Angew. Chem., Int. Ed. 2014, 53, 12064–12068. doi:10.1002/anie.201406450 |
| 27. | López-Mendoza, P.; Díaz, J. E.; Loaiza, A. E.; Miranda, L. D. Tetrahedron 2018, 74, 5494–5502. doi:10.1016/j.tet.2018.04.079 |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 62. | Devery, J. J., III; Nguyen, J. D.; Dai, C.; Stephenson, C. R. J. ACS Catal. 2016, 6, 5962–5967. doi:10.1021/acscatal.6b01914 |
| 63. | Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599 |
| 25. | Mestre, F.; Tailham, C.; Zard, S. Z. Heterocycles 1989, 28, 171–174. doi:10.3987/com-88-s77 |
| 26. | Delduc, P.; Tailhan, C.; Zard, S. Z. J. Chem. Soc., Chem. Commun. 1988, 308–310. doi:10.1039/c39880000308 |
| 56. | Coyle, J. D. Tetrahedron 1985, 41, 5393–5425. doi:10.1016/s0040-4020(01)91341-9 |
| 22. | Tazhe Veetil, A.; Šolomek, T.; Ngoy, B. P.; Pavlíková, N.; Heger, D.; Klán, P. J. Org. Chem. 2011, 76, 8232–8242. doi:10.1021/jo201385b |
| 23. | Ferjančić, Z.; Čeković, Ž.; Saičić, R. N. Tetrahedron Lett. 2000, 41, 2979–2982. doi:10.1016/s0040-4039(00)00286-0 |
| 24. | Maslak, V.; Čeković, Ž.; Saičić, R. N. Synlett 1998, 1435–1437. doi:10.1055/s-1998-1946 |
| 25. | Mestre, F.; Tailham, C.; Zard, S. Z. Heterocycles 1989, 28, 171–174. doi:10.3987/com-88-s77 |
| 26. | Delduc, P.; Tailhan, C.; Zard, S. Z. J. Chem. Soc., Chem. Commun. 1988, 308–310. doi:10.1039/c39880000308 |
| 38. | Ding, C.; Fan, C.; Jiang, G.; Pan, X.; Zhang, Z.; Zhu, J.; Zhu, X. Macromol. Rapid Commun. 2015, 36, 2181–2185. doi:10.1002/marc.201500427 |
| 39. | Shanmugam, S.; Xu, J.; Boyer, C. Macromolecules 2014, 47, 4930–4942. doi:10.1021/ma500842u |
| 40. | Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. J. Am. Chem. Soc. 2014, 136, 5508–5519. doi:10.1021/ja501745g |
| 41. | Phommalysack-Lovan, J.; Chu, Y.; Boyer, C.; Xu, J. Chem. Commun. 2018, 54, 6591–6606. doi:10.1039/c8cc02783h |
| 57. | Cismesia, M. A.; Yoon, T. P. Chem. Sci. 2015, 6, 5426–5434. doi:10.1039/c5sc02185e |
| 58. | Hatchard, C. G.; Parker, C. A. Proc. R. Soc. London, Ser. A 1956, 235, 518–536. doi:10.1098/rspa.1956.0102 |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 47. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 48. | Shen, T.; Zhao, Z.-G.; Yu, Q.; Xu, H.-J. J. Photochem. Photobiol., A 1989, 47, 203–212. doi:10.1016/1010-6030(89)87066-2 |
| 49. | Du, Y.; Pearson, R. M.; Lim, C.-H.; Sartor, S. M.; Ryan, M. D.; Yang, H.; Damrauer, N. H.; Miyake, G. M. Chem. – Eur. J. 2017, 23, 10962–10968. doi:10.1002/chem.201702926 |
| 50. | Singh, A.; Teegardin, K.; Kelly, M.; Prasad, K. S.; Krishnan, S.; Weaver, J. D. J. Organomet. Chem. 2015, 776, 51–59. doi:10.1016/j.jorganchem.2014.10.037 |
| 48. | Shen, T.; Zhao, Z.-G.; Yu, Q.; Xu, H.-J. J. Photochem. Photobiol., A 1989, 47, 203–212. doi:10.1016/1010-6030(89)87066-2 |
| 49. | Du, Y.; Pearson, R. M.; Lim, C.-H.; Sartor, S. M.; Ryan, M. D.; Yang, H.; Damrauer, N. H.; Miyake, G. M. Chem. – Eur. J. 2017, 23, 10962–10968. doi:10.1002/chem.201702926 |
| 53. | Teders, M.; Henkel, C.; Anhäuser, L.; Strieth-Kalthoff, F.; Gómez-Suárez, A.; Kleinmans, R.; Kahnt, A.; Rentmeister, A.; Guldi, D.; Glorius, F. Nat. Chem. 2018, 10, 981–988. doi:10.1038/s41557-018-0102-z |
| 53. | Teders, M.; Henkel, C.; Anhäuser, L.; Strieth-Kalthoff, F.; Gómez-Suárez, A.; Kleinmans, R.; Kahnt, A.; Rentmeister, A.; Guldi, D.; Glorius, F. Nat. Chem. 2018, 10, 981–988. doi:10.1038/s41557-018-0102-z |
| 54. | van As, D. J.; Connell, T. U.; Brzozowski, M.; Scully, A. D.; Polyzos, A. Org. Lett. 2018, 20, 905–908. doi:10.1021/acs.orglett.7b03565 |
| 55. | Ichimura, K.; Kobayashi, T.; King, K. A.; Watts, R. J. J. Phys. Chem. 1987, 91, 6104–6106. doi:10.1021/j100308a012 |
| 47. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 51. | Strieth-Kalthoff, F.; James, M. J.; Teders, M.; Pitzer, L.; Glorius, F. Chem. Soc. Rev. 2018, 47, 7190–7202. doi:10.1039/c8cs00054a |
| 52. | Xiao, W.-J.; Zhou, Q.-Q.; Zou, Y.-Q.; Lu, L.-Q. Angew. Chem., Int. Ed. 2018. doi:10.1002/anie.201803102 |
| 50. | Singh, A.; Teegardin, K.; Kelly, M.; Prasad, K. S.; Krishnan, S.; Weaver, J. D. J. Organomet. Chem. 2015, 776, 51–59. doi:10.1016/j.jorganchem.2014.10.037 |
| 46. | Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156–1163. doi:10.1021/acs.oprd.6b00101 |
| 47. | Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712–5719. doi:10.1021/cm051312+ |
| 50. | Singh, A.; Teegardin, K.; Kelly, M.; Prasad, K. S.; Krishnan, S.; Weaver, J. D. J. Organomet. Chem. 2015, 776, 51–59. doi:10.1016/j.jorganchem.2014.10.037 |
© 2018 Kaga et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)