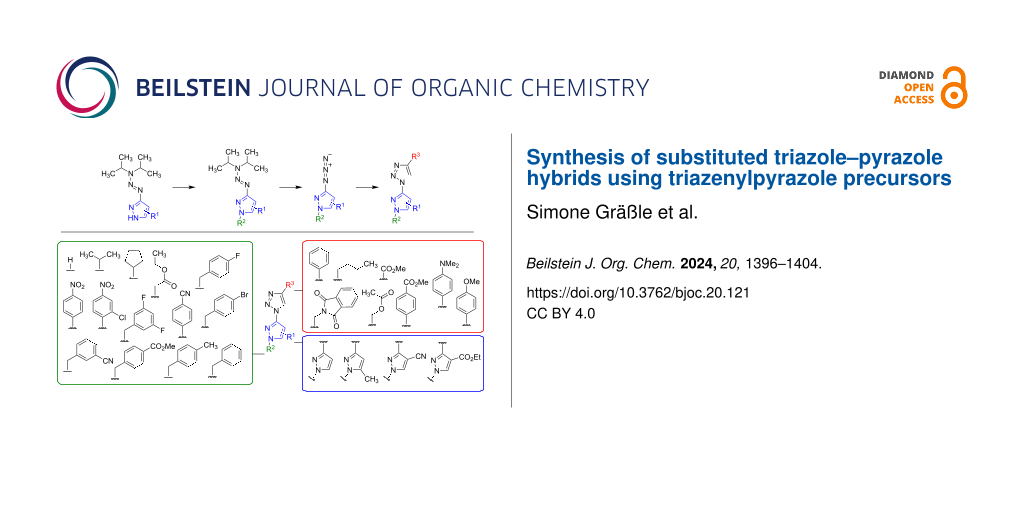
Abstract
A synthesis route to access triazole–pyrazole hybrids via triazenylpyrazoles was developed. Contrary to existing methods, this route allows the facile N-functionalization of the pyrazole before the attachment of the triazole unit via a copper-catalyzed azide–alkyne cycloaddition. The developed methodology was used to synthesize a library of over fifty new multi-substituted pyrazole–triazole hybrids. We also demonstrate a one-pot strategy that renders the isolation of potentially hazardous azides obsolete. In addition, the compatibility of the method with solid-phase synthesis is shown exemplarily.

Graphical Abstract
Introduction
Nitrogen-containing heterocycles are central scaffolds in medicinal chemistry and are incorporated in most small-molecule drugs [1,2]. We are interested in feasible strategies to synthesize nitrogen-rich heterocyclic scaffolds that can extend the currently available libraries with new drug-like molecules. Our past work on pyrazoles [3-6] and triazoles [7-11] motivated us to search for suitable and versatile strategies to explore access to triazole–pyrazole hybrids. Triazole–pyrazole hybrids, particularly non-fused heterocycles of this class, have not been investigated systematically. Selected known derivatives (Figure 1, 1–4) inhibit the serine-threonine kinase ERK3 [12] or the cholera-causing bacterium Vibrio cholerae [13], show antimicrobial properties [14], and can act as P2X7 antagonists, a receptor involved in neuroinflammation and depression [15].
![[1860-5397-20-121-1]](/bjoc/content/figures/1860-5397-20-121-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active pyrazole–triazole hybrids 1–4: inhibitory effect on cholera bacteria [13], antimicrobial properties [14], P2X7 antagonists (depression) [15] and ERK3 inhibition [12].
Figure 1: Biologically active pyrazole–triazole hybrids 1–4: inhibitory effect on cholera bacteria [13], antimicr...
Pyrazolyltriazoles are most easily obtained via the copper-catalyzed azide–alkyne cycloaddition (CuAAC) from pyrazolyl azides (7 and 8). These are usually accessed from the respective amines or organohalides (5 and 6, Scheme 1) [14,16-18]. Few examples of triazole–pyrazole hybrids, such as 13, have also been synthesized through a modified Sakai reaction [19], a reaction cascade involving the elimination of an azole [20] or in the n-butyllithium-mediated reaction with alkyl halides [21]. So far, the literature-reported methods are most often limited to N-unsubstituted pyrazoles or triazoles and pyrazoles being fused to a second (hetero)cycle; the synthesis of promising multi-substituted structures such as 1 has not yet been described systematically.
![[1860-5397-20-121-i1]](/bjoc/content/inline/1860-5397-20-121-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Literature-reported synthetic routes to pyrazole–triazole hybrids: synthesis of azides 7 or 8 from amines and organohalides and subsequent CuAAC to larger heterocyclic systems 9 or non-substituted amine products 10; Sakai reaction of α-ketoacetal 11 for the synthesis of N-substituted derivative 13 [14,16-19].
Scheme 1: Literature-reported synthetic routes to pyrazole–triazole hybrids: synthesis of azides 7 or 8 from ...
Results and Discussion
Triazenes have previously been established as versatile intermediates and linkers for conventional and solid-phase synthesis [22-25] that can be considered as protected diazonium salts [3]. According to the previous work [3], triazenylpyrazoles could serve as azide sources and thus as building blocks for synthesizing pyrazolyltriazoles by CuAAC reactions. To find a feasible approach to pyrazolyltriazoles of type 1 with a highly substituted scaffold, we decided to explore the benefits of a modification of the triazene-protected pyrazole core. In the next step, a cycloaddition of the gained synthesized azidopyrazoles with different alkynes was to be conducted.
The 3-(3,3-diisopropyltriaz-1-en-1-yl)-1H-pyrazole precursors 15a–d were synthesized according to previously reported procedures [3,26,27] via the generation of a diazonium salt from aminopyrazoles 14a–d followed by the addition of diisopropylamine, either in a one-pot synthesis or in two consecutive steps (Table 1). Subsequently, different aliphatic and aromatic substituents were attached to the pyrazole nitrogen by nucleophilic substitution with suitable organohalides 16 and cesium carbonate [3]. Due to the pyrazole tautomerism [28], the formation of two possible regioisomers, 17 and 18, was anticipated and could be confirmed experimentally. Depending on the employed halide 16, the distribution of the obtained products varied. A considerable excess of the dominating isomer with yields of up to 70% could be obtained in some cases (see 17f or 17m), whereas the isomers were isolated in a 1:1 ratio for compounds 17c or 17h. A strong trend towards regioisomer 17 as the main product was observed for substituted phenyl residues, presumably due to the higher steric hindrance (see 17e–g). The results for benzylic residues differed depending on the benzylic residue's functional groups and the pyrazole substitution pattern. For starting materials 15a and 15d, an excess of product 17 was usually observed. With the ester-functionalized triazene 15c and m-substituted benzylic reagents, regioisomer 18 was the predominant product (see 18i and 18j). In total, 13 groups could be attached to the different triazenylpyrazoles, yielding 18 products (see Table 1).
Table 1: Synthesis of triazenylpyrazoles 15a–d and functionalization to N-substituted triazenylpyrazoles 17a–r and 18a–r. Conditions i: 1) BF3·OEt2, isoamyl nitrite, THF, −20 °C, 1 h, 2) diisopropylamine, THF/pyridine/acetonitrile, −20 °C to 21 °C, 17 h; Conditions ii: 1) HClaq (6 M), NaNO2, 0 °C to 5 °C, 1–2 h, 2) diisopropylamine, 0 °C to 21 °C, 16 h. X = F, Br, I.
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-121-i6.svg?max-width=637&scale=1.0)
|
|||||
| R1 | R2 | Compound | Yield 17 [%] | Compound | Yield 18 [%] |
| H | C | 17a | 59 | 18a | 18 |
| H | D | 17b | 34 | 18b | 28 |
| H | M | 17c | 40 | 18c | 40 |
| 5-Me | C | 17d | 23 | 18d | 29 |
| 5-Me | J | 17e | 58 | 18e | 12 |
| 5-Me | K | 17f | 64 | 18f | 19 |
| 5-Me | L | 17g | 61 | 18g | 9 |
| 4-CO2Et | C | 17h | 44 | 18h | 40 |
| 4-CO2Et | H | 17i | 37 | 18i | 63 |
| 4-CO2Et | I | 17j | 35 | 18j | 65 |
| 4-CO2Et | D | 17k | 41 | 18k | 59 |
| 4-CN | A | 17l | 35 | 18l | 58 |
| 4-CN | B | 17m | 70 | 18m | 22 |
| 4-CN | C | 17n | 58 | 18n | 39 |
| 4-CN | H | 17o | 51 | 18o | 46 |
| 4-CN | E | 17p | 59 | 18p | 40 |
| 4-CN | F | 17q | 53 | 18q | 45 |
| 4-CN | G | 17r | 54 | 18r | 41 |
In analogy to reported procedures for cleavage of polymer-bound triazenes [23], we attempted to develop the first protocol for synthesizing pyrazolyl azides 19 from triazenylpyrazoles. Initial experiments with TFA and trimethylsilyl azide at 0–25 °C in DCM failed for 4-substituted pyrazoles; the formation of the target products was only observed when 5-methylpyrazoles such as 15b were used. Therefore, a modified procedure was applied, heating the triazenes to 50 °C. This optimization allowed for the isolation of the corresponding azides 19a–v in yields of 51% to quantitative (Scheme 2), usually with durations of 3–16 h. Longer reaction times were necessary for some CN-substituted triazene derivatives (19o, 19q, 19s, 19v), especially in combination with electron-withdrawing functional groups. The developed procedure could only be used to convert isomer 17. Triazene compounds with the regioisomeric form 18 could not be reacted (see Supporting Information File 1, Scheme S1) even after extended reaction times, only starting material was reisolated, presumably due to the increased stability of isomer 18 towards acids. This corresponds with the results for the previously reported triazene cleavage to diazonium intermediates and subsequent cyclization to triazine derivatives [3].
![[1860-5397-20-121-i2]](/bjoc/content/inline/1860-5397-20-121-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of pyrazolyl azides 19a–v via cleavage of the protecting triazene moiety. For compounds 19a–h, 19p, 19r, 19t and 19u, reaction times of 3–16 h were suitable. The reactions to 19o and 19v were stirred for 3 d; for 19q and 19s, reaction times of 7 d were necessary.
Scheme 2: Synthesis of pyrazolyl azides 19a–v via cleavage of the protecting triazene moiety. For compounds 1...
In the next step, the obtained pyrazolyl azides were reacted with different aromatic and aliphatic alkynes 20a–h in a copper-catalyzed azide–alkyne cycloaddition (CuAAC). All attempted reactions could be conducted under standard conditions using copper sulfate and sodium ascorbate in THF/water (depicted in Scheme 3 and Figure 2). For selected derivatives, 21sd and 21vg, crystals suitable for single-crystal X-ray diffraction could be obtained and confirmed the product structure with the presumed regioisomer (Scheme 3).
![[1860-5397-20-121-i3]](/bjoc/content/inline/1860-5397-20-121-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of pyrazole–triazole hybrids via CuAAC and ORTEP diagrams of triazole products 21sd and 21vg with the thermal ellipsoids shown at 50% probability. NaAsc = sodium ascorbate.
Scheme 3: Synthesis of pyrazole–triazole hybrids via CuAAC and ORTEP diagrams of triazole products 21sd and 2...
A library of over 50 triazole products 21aa–vg was successfully synthesized with yields ranging from 28% to quantitative, combining four different pyrazole-carbon substitutions and 14 pyrazole-nitrogen substitutions with eight different residues on the to-be-formed triazole (see Figure 2). It could be observed that the cycloaddition proceeds least efficiently with pyrazoles that are not substituted on the nitrogen. The reaction of pyrazolyl azides 19e and 19j with phenylacetylene gave the products 21ed and 21jd with yields of 57% and 28%, whereas substituted derivatives (e.g., 19g or 19n) resulted in yields of over 90% (21gd or 21nd) using the same alkyne. The different substitution patterns on the 4- or 5-position of the pyrazole (R1) do not clearly influence the reaction's efficiency. Although the reactions of ethyl 3-azido-1H-pyrazole-4-carboxylate (19j) resulted in lower yields of the triazole products 21ja–jh compared to pyrazolyl azides 19a, 19e, and 19o, this trend is not continued in the results of the N-substituted carboxylate derivatives 19k–n. The effect of the alkyne depends on the substitutions on the pyrazole, and no general trend is visible – reactions with electron-poor, electron-rich as well as sterically demanding alkynes give high product yields, depending on the respective pyrazole. The functionalization of the NH-unsubstituted derivative 21jd via copper-catalyzed cross-coupling [29] with an electron-rich aryl substituent was exemplarily conducted to further expand the scope of possible products (see Scheme S2, Supporting Information File 1).
![[1860-5397-20-121-2]](/bjoc/content/figures/1860-5397-20-121-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesized triazole–pyrazole hybrids 21aa–vg.
Figure 2: Synthesized triazole–pyrazole hybrids 21aa–vg.
We also investigated the scope and limitations of a one-pot reaction for the triazene cleavage and subsequent CuAAC with the model compound 17e (see Scheme 4). When conducting the two reaction steps back-to-back in a one-pot setup, a decrease in yield from 86% over two steps (96% and 90%) to 59% of impure product was observed. This is presumably caused by incomplete conversion of the in situ-generated alkyne to the triazole. The decrease of TFA/TMS-N3 in the reaction or the addition of an increased amount of alkyne further deteriorated the results. Therefore, we introduced a straightforward evaporation step after completion of the triazene cleavage to remove the residual reagents. The final product 21gd could be isolated in quantitative yield with this technique, avoiding additional purification steps for the azide intermediate without any losses in product formation.
![[1860-5397-20-121-i4]](/bjoc/content/inline/1860-5397-20-121-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: One-pot synthesis of triazole–pyrazole hybrid 21gd. aOne-pot setup yielded 21gd with unknown impurities; bwith an additional evaporation step after the triazene cleavage, a quantitative yield of the target product 21gd was achieved.
Scheme 4: One-pot synthesis of triazole–pyrazole hybrid 21gd. aOne-pot setup yielded 21gd with unknown impuri...
The developed procedure was exemplarily transferred to solid-phase synthesis. In quantitative yields, 5-methyl-1H-pyrazol-3-amine (14b) was immobilized on benzylamine resin 22 (Scheme 5). For this purpose, a diazonium intermediate was generated from the pyrazoloamine with BF3∙Et2O and isoamyl nitrite accordingly to the liquid phase synthesis of 15b. The subsequent functionalization of resin 23 to the phenyl-substituted derivative 25 was carried out using the nucleophilic substitution procedure reported above with yields of 63–76%. The anticipated formation of a second regioisomer could not be confirmed due to the limited analytical methods available for compounds on solid supports. The cleavage to obtain azidopyrazole 19g was achieved with a total yield of 37% over all steps, comparable to the total yield of 45% for the stepwise synthesis in the liquid phase. This indicates a material loss due to the non-reactive regioisomer formation in the previous step and a non-quantitative cleavage process. In analogy to the one-pot experiments in solution, a one-pot cleavage from the resin combined with the CuAAC reaction to the triazole–pyrazole hybrid was conducted exemplarily and gave the target product 21gd in 30% yield.
![[1860-5397-20-121-i5]](/bjoc/content/inline/1860-5397-20-121-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Solid-phase synthesis of azidopyrazole 19g and triazole–pyrazole hybrid 21gd by immobilization of aminopyrazole 14b on benzylamine-substituted Merrifield resin 22, NH-functionalization and cleavage. Reaction conditions: a) BF3∙Et2O, isoamyl nitrite, THF/pyridine (9:1), −20 °C to 21 °C, 12 h, b) Cs2CO3, DMSO, 120 °C, 2–3 d, c) TFA, TMS-N3, DCM, 25 °C, 12 h, d) 1. TFA, TMS-N3, DCM, 0–50 °C, 12 h; 2. alkyne, THF/H2O, CuSO4, sodium ascorbate, 16 h, 50 °C.
Scheme 5: Solid-phase synthesis of azidopyrazole 19g and triazole–pyrazole hybrid 21gd by immobilization of a...
The solid-phase reaction route allows for roughly equally high overall yields compared to the solution synthesis. It offers the additional benefits of chemistry on solid support: straightforward purification of the resin-bound intermediates by washing steps and a high throughput that allows for faster derivatization. Further research is necessary to establish a protocol for the cleavage of 4-substituted pyrazoles, as the corresponding azides analogous to 19j–v could not be obtained from immobilized triazene precursors.
Conclusion
A synthesis route to access substituted triazole–pyrazole hybrids from triazenylpyrazoles has been established and applied to obtain a library of over 50 new triazole–pyrazole hybrids. The selective N-functionalization of the triazene-protected pyrazoles was conducted, and the cleavage of triazenylpyrazoles to the corresponding azides was described for the first time with regioisomer 17, whereas regioisomer 18 is acid-insensitive and cannot be converted. The azides were reacted to the respective triazole product in a CuAAC reaction; this step could also successfully be conducted in a sequential one-pot approach from the triazenylpyrazole precursor. The developed protocol was adapted for solid-phase synthesis to demonstrate the applicability of triazenylpyrazoles as immobilized building blocks.
Supporting Information
The Supporting Information covers detailed material on the conducted experiments and their results. All experimental details, including the analytical description of the obtained target compounds and byproducts, are available in the Supporting Information. Data that refers to the experiments described herein were submitted to the repository chemotion (https://www.chemotion-repository.net/). All DOIs minted for the data are linked in Supporting Information File 1 and the NMR spectra are given in Supporting Information File 2. Information on the availability of the data and the physical material of the target compounds is added to Supporting Information File 3. New data obtained in this study is assigned to the collection embargo number SGV_2021-06-02 (https://dx.doi.org/10.14272/collection/SGV_2021-06-02) [30]. The material obtained in this study was submitted to the Molecule Archive at KIT and can be requested from there (https://compound-platform.eu/home). CCDC 2308695 (18n), 2308696 (21vg) and 2309318 (21sd) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 15.4 MB | Download |
| Supporting Information File 3: Information on the availability of the data and the physical material of the target compounds. | ||
| Format: PDF | Size: 872.5 KB | Download |
Funding
This work was supported by the Helmholtz program Information. We acknowledge support by Deutsche Forschungsgemeinschaft for the DFG-core facility Molecule Archive, to which all target compounds were registered for further re-use (DFG project number: 284178167). We thank NFDI4Chem (project number: 441958208) for providing the necessary infrastructure to process, store, and share the research data gained in this study.
Data Availability Statement
The data generated and analyzed during this study is openly available in the Chemotion repository at https://doi.org/10.14272/collection/SGV_2021-06-02. Crystal structures are made available via the CCDC.
References
-
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g
Return to citation in text: [1] -
Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Wezeman, T.; Comas-Barceló, J.; Nieger, M.; Harrity, J. P. A.; Bräse, S. Org. Biomol. Chem. 2017, 15, 1575–1579. doi:10.1039/c6ob02518h
Return to citation in text: [1] -
Rüger, A. J.; Nieger, M.; Bräse, S. Tetrahedron 2012, 68, 8823–8829. doi:10.1016/j.tet.2012.07.069
Return to citation in text: [1] -
Busch, M.; Cayir, M.; Nieger, M.; Thiel, W. R.; Bräse, S. Eur. J. Org. Chem. 2013, 6108–6123. doi:10.1002/ejoc.201300508
Return to citation in text: [1] -
Althuon, D.; Rönicke, F.; Fürniss, D.; Quan, J.; Wellhöfer, I.; Jung, N.; Schepers, U.; Bräse, S. Org. Biomol. Chem. 2015, 13, 4226–4230. doi:10.1039/c5ob00250h
Return to citation in text: [1] -
Holzhauer, L.; Liagre, C.; Fuhr, O.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2022, 18, 1088–1099. doi:10.3762/bjoc.18.111
Return to citation in text: [1] -
Plietzsch, O.; Schilling, C. I.; Tolev, M.; Nieger, M.; Richert, C.; Muller, T.; Bräse, S. Org. Biomol. Chem. 2009, 7, 4734–4743. doi:10.1039/b912189g
Return to citation in text: [1] -
Zimmermann, V.; Avemaria, F.; Bräse, S. J. Comb. Chem. 2007, 9, 200–203. doi:10.1021/cc060116c
Return to citation in text: [1] -
Vidyakina, A. A.; Shtyrov, A. A.; Ryazantsev, M. N.; Khlebnikov, A. F.; Kolesnikov, I. E.; Sharoyko, V. V.; Spiridonova, D. V.; Balova, I. A.; Bräse, S.; Danilkina, N. A. Chem. – Eur. J. 2023, 29, e202300540. doi:10.1002/chem.202300540
Return to citation in text: [1] -
Grädler, U.; Busch, M.; Leuthner, B.; Raba, M.; Burgdorf, L.; Lehmann, M.; Linde, N.; Esdar, C. Bioorg. Med. Chem. Lett. 2020, 30, 127551. doi:10.1016/j.bmcl.2020.127551
Return to citation in text: [1] [2] -
Bolger, G.; Roy, S.; Zapol'skii, V. A.; Kaufmann, D. E.; Schnürch, M.; Mihovilovic, M. D.; Nandy, R. K.; Tegge, W. J. Med. Microbiol. 2016, 65, 678–687. doi:10.1099/jmm.0.000276
Return to citation in text: [1] [2] -
Nagender, P.; Malla Reddy, G.; Naresh Kumar, R.; Poornachandra, Y.; Ganesh Kumar, C.; Narsaiah, B. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. doi:10.1016/j.bmcl.2014.04.084
Return to citation in text: [1] [2] [3] [4] -
Chrovian, C. C.; Soyode-Johnson, A.; Peterson, A. A.; Gelin, C. F.; Deng, X.; Dvorak, C. A.; Carruthers, N. I.; Lord, B.; Fraser, I.; Aluisio, L.; Coe, K. J.; Scott, B.; Koudriakova, T.; Schoetens, F.; Sepassi, K.; Gallacher, D. J.; Bhattacharya, A.; Letavic, M. A. J. Med. Chem. 2018, 61, 207–223. doi:10.1021/acs.jmedchem.7b01279
Return to citation in text: [1] [2] -
Dalinger, A. I.; Medved’ko, A. V.; Balalaeva, A. I.; Vatsadze, I. А.; Dalinger, I. L.; Vatsadze, S. Z. Chem. Heterocycl. Compd. 2020, 56, 180–191. doi:10.1007/s10593-020-02643-2
Return to citation in text: [1] [2] -
Clarke, D.; Mares, R. W.; McNab, H. J. Chem. Soc., Perkin Trans. 1 1997, 1799–1804. doi:10.1039/a700421d
Return to citation in text: [1] [2] -
Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Org. Biomol. Chem. 2009, 7, 3421–3429. doi:10.1039/b906482f
Return to citation in text: [1] [2] -
Zehnder, L. R.; Hawkins, J. M.; Sutton, S. C. Synlett 2020, 31, 175–178. doi:10.1055/s-0039-1691526
Return to citation in text: [1] [2] -
Zapol’skii, V. A.; Berneburg, I.; Bilitewski, U.; Dillenberger, M.; Becker, K.; Jungwirth, S.; Shekhar, A.; Krueger, B.; Kaufmann, D. E. Beilstein J. Org. Chem. 2022, 18, 524–532. doi:10.3762/bjoc.18.54
Return to citation in text: [1] -
Kim, T.; Kim, K. J. Heterocycl. Chem. 2010, 47, 98–111. doi:10.1002/jhet.275
Return to citation in text: [1] -
Bräse, S.; Enders, D.; Köbberling, J.; Avemaria, F. Angew. Chem., Int. Ed. 1998, 37, 3413–3415. doi:10.1002/(sici)1521-3773(19981231)37:24<3413::aid-anie3413>3.0.co;2-k
Return to citation in text: [1] -
Avemaria, F.; Zimmermann, V.; Bräse, S. Synlett 2004, 1163–1166. doi:10.1055/s-2004-82298
Return to citation in text: [1] [2] -
Garcia, A. M.; Jung, N.; Gil, C.; Nieger, M.; Bräse, S. RSC Adv. 2015, 5, 65540–65545. doi:10.1039/c5ra09705c
Return to citation in text: [1] -
Reingruber, R.; Vanderheiden, S.; Muller, T.; Nieger, M.; Es-Sayed, M.; Bräse, S. Tetrahedron Lett. 2009, 50, 3439–3442. doi:10.1016/j.tetlet.2009.02.184
Return to citation in text: [1] -
Larsen, J. S.; Zahran, M. A.; Pedersen, E. B.; Nielsen, C. Monatsh. Chem. 1999, 130, 1167–1173. doi:10.1007/pl00010295
Return to citation in text: [1] -
Döbele, M.; Vanderheiden, S.; Jung, N.; Bräse, S. Angew. Chem., Int. Ed. 2010, 49, 5986–5988. doi:10.1002/anie.201001507
Return to citation in text: [1] -
Aguilar-Parrilla, F.; Cativiela, C.; de Villegas, M. D. D.; Elguero, J.; Foces-Foces, C.; Laureiro, J. I. G.; Cano, F. H.; Limbach, H.-H.; Smith, J. A. S.; Toiron, C. J. Chem. Soc., Perkin Trans. 2 1992, 1737. doi:10.1039/p29920001737
Return to citation in text: [1] -
Wippert, N. A.; Jung, N.; Bräse, S. ACS Comb. Sci. 2019, 21, 568–572. doi:10.1021/acscombsci.9b00096
Return to citation in text: [1] -
Graessle, S.; Wippert, N.; Holzhauer, L. Chemotion Repository. 2024; https://www.chemotion-repository.net/home/publications/collections/3613. doi:10.14272/collection/sgv_2021-06-02
Return to citation in text: [1]
| 1. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 2. | Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g |
| 13. | Bolger, G.; Roy, S.; Zapol'skii, V. A.; Kaufmann, D. E.; Schnürch, M.; Mihovilovic, M. D.; Nandy, R. K.; Tegge, W. J. Med. Microbiol. 2016, 65, 678–687. doi:10.1099/jmm.0.000276 |
| 12. | Grädler, U.; Busch, M.; Leuthner, B.; Raba, M.; Burgdorf, L.; Lehmann, M.; Linde, N.; Esdar, C. Bioorg. Med. Chem. Lett. 2020, 30, 127551. doi:10.1016/j.bmcl.2020.127551 |
| 14. | Nagender, P.; Malla Reddy, G.; Naresh Kumar, R.; Poornachandra, Y.; Ganesh Kumar, C.; Narsaiah, B. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. doi:10.1016/j.bmcl.2014.04.084 |
| 16. | Dalinger, A. I.; Medved’ko, A. V.; Balalaeva, A. I.; Vatsadze, I. А.; Dalinger, I. L.; Vatsadze, S. Z. Chem. Heterocycl. Compd. 2020, 56, 180–191. doi:10.1007/s10593-020-02643-2 |
| 17. | Clarke, D.; Mares, R. W.; McNab, H. J. Chem. Soc., Perkin Trans. 1 1997, 1799–1804. doi:10.1039/a700421d |
| 18. | Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Org. Biomol. Chem. 2009, 7, 3421–3429. doi:10.1039/b906482f |
| 19. | Zehnder, L. R.; Hawkins, J. M.; Sutton, S. C. Synlett 2020, 31, 175–178. doi:10.1055/s-0039-1691526 |
| 7. | Althuon, D.; Rönicke, F.; Fürniss, D.; Quan, J.; Wellhöfer, I.; Jung, N.; Schepers, U.; Bräse, S. Org. Biomol. Chem. 2015, 13, 4226–4230. doi:10.1039/c5ob00250h |
| 8. | Holzhauer, L.; Liagre, C.; Fuhr, O.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2022, 18, 1088–1099. doi:10.3762/bjoc.18.111 |
| 9. | Plietzsch, O.; Schilling, C. I.; Tolev, M.; Nieger, M.; Richert, C.; Muller, T.; Bräse, S. Org. Biomol. Chem. 2009, 7, 4734–4743. doi:10.1039/b912189g |
| 10. | Zimmermann, V.; Avemaria, F.; Bräse, S. J. Comb. Chem. 2007, 9, 200–203. doi:10.1021/cc060116c |
| 11. | Vidyakina, A. A.; Shtyrov, A. A.; Ryazantsev, M. N.; Khlebnikov, A. F.; Kolesnikov, I. E.; Sharoyko, V. V.; Spiridonova, D. V.; Balova, I. A.; Bräse, S.; Danilkina, N. A. Chem. – Eur. J. 2023, 29, e202300540. doi:10.1002/chem.202300540 |
| 19. | Zehnder, L. R.; Hawkins, J. M.; Sutton, S. C. Synlett 2020, 31, 175–178. doi:10.1055/s-0039-1691526 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
| 4. | Wezeman, T.; Comas-Barceló, J.; Nieger, M.; Harrity, J. P. A.; Bräse, S. Org. Biomol. Chem. 2017, 15, 1575–1579. doi:10.1039/c6ob02518h |
| 5. | Rüger, A. J.; Nieger, M.; Bräse, S. Tetrahedron 2012, 68, 8823–8829. doi:10.1016/j.tet.2012.07.069 |
| 6. | Busch, M.; Cayir, M.; Nieger, M.; Thiel, W. R.; Bräse, S. Eur. J. Org. Chem. 2013, 6108–6123. doi:10.1002/ejoc.201300508 |
| 20. | Zapol’skii, V. A.; Berneburg, I.; Bilitewski, U.; Dillenberger, M.; Becker, K.; Jungwirth, S.; Shekhar, A.; Krueger, B.; Kaufmann, D. E. Beilstein J. Org. Chem. 2022, 18, 524–532. doi:10.3762/bjoc.18.54 |
| 14. | Nagender, P.; Malla Reddy, G.; Naresh Kumar, R.; Poornachandra, Y.; Ganesh Kumar, C.; Narsaiah, B. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. doi:10.1016/j.bmcl.2014.04.084 |
| 12. | Grädler, U.; Busch, M.; Leuthner, B.; Raba, M.; Burgdorf, L.; Lehmann, M.; Linde, N.; Esdar, C. Bioorg. Med. Chem. Lett. 2020, 30, 127551. doi:10.1016/j.bmcl.2020.127551 |
| 13. | Bolger, G.; Roy, S.; Zapol'skii, V. A.; Kaufmann, D. E.; Schnürch, M.; Mihovilovic, M. D.; Nandy, R. K.; Tegge, W. J. Med. Microbiol. 2016, 65, 678–687. doi:10.1099/jmm.0.000276 |
| 14. | Nagender, P.; Malla Reddy, G.; Naresh Kumar, R.; Poornachandra, Y.; Ganesh Kumar, C.; Narsaiah, B. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. doi:10.1016/j.bmcl.2014.04.084 |
| 16. | Dalinger, A. I.; Medved’ko, A. V.; Balalaeva, A. I.; Vatsadze, I. А.; Dalinger, I. L.; Vatsadze, S. Z. Chem. Heterocycl. Compd. 2020, 56, 180–191. doi:10.1007/s10593-020-02643-2 |
| 17. | Clarke, D.; Mares, R. W.; McNab, H. J. Chem. Soc., Perkin Trans. 1 1997, 1799–1804. doi:10.1039/a700421d |
| 18. | Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Org. Biomol. Chem. 2009, 7, 3421–3429. doi:10.1039/b906482f |
| 15. | Chrovian, C. C.; Soyode-Johnson, A.; Peterson, A. A.; Gelin, C. F.; Deng, X.; Dvorak, C. A.; Carruthers, N. I.; Lord, B.; Fraser, I.; Aluisio, L.; Coe, K. J.; Scott, B.; Koudriakova, T.; Schoetens, F.; Sepassi, K.; Gallacher, D. J.; Bhattacharya, A.; Letavic, M. A. J. Med. Chem. 2018, 61, 207–223. doi:10.1021/acs.jmedchem.7b01279 |
| 14. | Nagender, P.; Malla Reddy, G.; Naresh Kumar, R.; Poornachandra, Y.; Ganesh Kumar, C.; Narsaiah, B. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. doi:10.1016/j.bmcl.2014.04.084 |
| 15. | Chrovian, C. C.; Soyode-Johnson, A.; Peterson, A. A.; Gelin, C. F.; Deng, X.; Dvorak, C. A.; Carruthers, N. I.; Lord, B.; Fraser, I.; Aluisio, L.; Coe, K. J.; Scott, B.; Koudriakova, T.; Schoetens, F.; Sepassi, K.; Gallacher, D. J.; Bhattacharya, A.; Letavic, M. A. J. Med. Chem. 2018, 61, 207–223. doi:10.1021/acs.jmedchem.7b01279 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
| 22. | Bräse, S.; Enders, D.; Köbberling, J.; Avemaria, F. Angew. Chem., Int. Ed. 1998, 37, 3413–3415. doi:10.1002/(sici)1521-3773(19981231)37:24<3413::aid-anie3413>3.0.co;2-k |
| 23. | Avemaria, F.; Zimmermann, V.; Bräse, S. Synlett 2004, 1163–1166. doi:10.1055/s-2004-82298 |
| 24. | Garcia, A. M.; Jung, N.; Gil, C.; Nieger, M.; Bräse, S. RSC Adv. 2015, 5, 65540–65545. doi:10.1039/c5ra09705c |
| 25. | Reingruber, R.; Vanderheiden, S.; Muller, T.; Nieger, M.; Es-Sayed, M.; Bräse, S. Tetrahedron Lett. 2009, 50, 3439–3442. doi:10.1016/j.tetlet.2009.02.184 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
| 30. | Graessle, S.; Wippert, N.; Holzhauer, L. Chemotion Repository. 2024; https://www.chemotion-repository.net/home/publications/collections/3613. doi:10.14272/collection/sgv_2021-06-02 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
| 29. | Wippert, N. A.; Jung, N.; Bräse, S. ACS Comb. Sci. 2019, 21, 568–572. doi:10.1021/acscombsci.9b00096 |
| 28. | Aguilar-Parrilla, F.; Cativiela, C.; de Villegas, M. D. D.; Elguero, J.; Foces-Foces, C.; Laureiro, J. I. G.; Cano, F. H.; Limbach, H.-H.; Smith, J. A. S.; Toiron, C. J. Chem. Soc., Perkin Trans. 2 1992, 1737. doi:10.1039/p29920001737 |
| 23. | Avemaria, F.; Zimmermann, V.; Bräse, S. Synlett 2004, 1163–1166. doi:10.1055/s-2004-82298 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
| 26. | Larsen, J. S.; Zahran, M. A.; Pedersen, E. B.; Nielsen, C. Monatsh. Chem. 1999, 130, 1167–1173. doi:10.1007/pl00010295 |
| 27. | Döbele, M.; Vanderheiden, S.; Jung, N.; Bräse, S. Angew. Chem., Int. Ed. 2010, 49, 5986–5988. doi:10.1002/anie.201001507 |
| 3. | Wippert, N.; Nieger, M.; Herlan, C.; Jung, N.; Bräse, S. Beilstein J. Org. Chem. 2021, 17, 2773–2780. doi:10.3762/bjoc.17.187 |
© 2024 Gräßle et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.