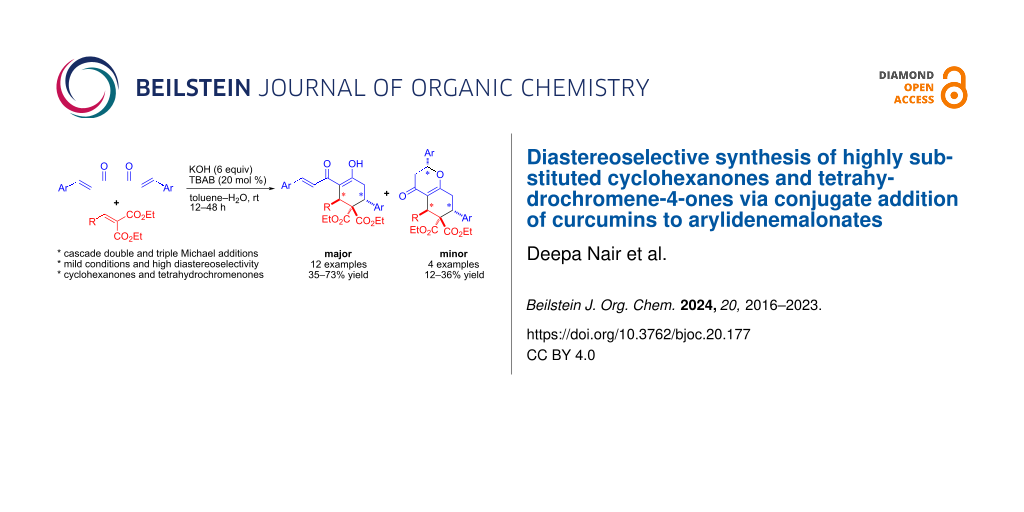
Abstract
A cascade inter–intramolecular double Michael strategy for the synthesis of highly functionalized cyclohexanones from curcumins and arylidenemalonates is reported. This strategy works in the presence of aqueous KOH using TBAB as a suitable phase transfer catalyst at room temperature. The functionalized cyclohexanones are formed as major products in moderate to excellent yields with complete diastereoselectivity in most cases. A triple Michael adduct, tetrahydrochromen-4-one, is also formed as a side product in a few cases with excellent diastereoselectivity.

Graphical Abstract
Introduction
There is considerable interest in the stereoselective synthesis of the cyclohexanone skeleton as it constitutes the core structure in many natural products and pharmaceutical drugs [1,2]. Garsubellin A with a cyclohexanone skeleton is a potent inducer of choline acetyltransferase (ChAT) and could be used for the treatment of Alzheimer's disease [3,4]. Likewise, RL91 and BHMPC are active for selective cell growth inhibition of the resistant lines (Figure 1) [5,6]. Their synthesis mainly involves a cascade Michael–aldol reaction between enones and suitable Michael donors such as β-ketosulfones, β-diketones, or double Michael addition of γ,δ-unsaturated-β-keto esters or Nazarov reagents with suitable acceptors such as nitroalkenes or alkylideneazalactones [7-10].
![[1860-5397-20-177-1]](/bjoc/content/figures/1860-5397-20-177-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active derivatives of cyclohexanones.
Figure 1: Biologically active derivatives of cyclohexanones.
From the perspective of an active methylene containing organic moiety, curcumin and its analogs serve as inexpensive and readily available starting materials for cascade/domino reactions. Curcumin is the key component present in turmeric and is responsible for its various biological activities. Turmeric has been used in many forms for potential health benefits and as a traditional spice for centuries [11]. Curcumin exhibits diverse biological properties such as anticancer, anti-HIV protease, anti-ageing, anti-inflammatory and anti-oxidant, to name a few [12-23]. Considering the fact that curcumin’s uniqueness is associated with its ability to function as a multifunctional substrate in various organic reactions, especially as a Michael donor at the central methylene carbon, and Michael acceptor at the enone vinyl carbon. Therefore, it would be interesting to develop novel methodologies using curcumin and its non-natural analogs as key starting materials [24]. Because of its multifaceted reactive site, curcumin showcases its Michael donor–acceptor ability in different ways, such as simple Michael addition, [4 + 2] annulation, Michael addition followed by cyclization or one-pot multicomponent reactions (MCR), etc. (Scheme 1) [25]. In 2011, our group reported the reactivity of curcumin as a Michael donor–acceptor with nitroalkenes, resulting in multi-substituted cyclohexanones through a cascade inter–intramolecular double Michael addition process with high diastereoselectivity [26,27]. Subsequently, the enantioselective versions of the above reaction and a similar diastereoselective cascade Michael addition–cyclization of curcumins with chalcones to synthesize functionalized cyclohexanones have been reported [28,29]. On the other hand, diastereoselective cascade Michael addition–cyclization of curcumins with α-bromonitroalkenes and α-halodicyclopentadienones afforded functionalized dihyrofurans [26,30]. The reaction of curcumins with azodicarboxylates, Morita–Baylis–Hillman derived nitroallylic acetates and α-hydrazinonitroalkenes were also investigated [29,31,32]. Very recently, our group has exploited 3-olefinic oxindoles and nitrochromenes to unfold the reactivities of curcumins [33,34].
![[1860-5397-20-177-i1]](/bjoc/content/inline/1860-5397-20-177-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Michael donor–acceptor reactivity of curcumin: previous vs present work.
Scheme 1: The Michael donor–acceptor reactivity of curcumin: previous vs present work.
Other groups have also investigated the Michael donor–acceptor reactivity of curcumins [25]. For instance, a quinine-thiourea catalyzed Michael addition of curcumins to nitroalkenes reported by Ye et al. stopped at the single Michael addition stage [35]. In the subsequent year, Yan et al. demonstrated a cascade triple-Michael (Michael/Michael/oxa-Michael) reaction between curcumins and isatylidene malononitriles, giving spiro-oxindoles in excellent yields and diastereoselectivities [36]. Sahu et al. introduced a one-pot multicomponent reaction of curcumin, arylaldehyde and 2-aminobenzothiazole to synthesize functionalized pyrimidobenzothiazoles [37]. An in situ generated conjugated α-cyanoester/malononitrile has been successfully employed as substrate in the double Michael reaction with curcumins by Lalitha et al. [38]. An organocatalytic cascade double Michael reaction between curcumins and 2-arylidene-1,3-indandiones was reported by Zhang and co-workers using quinine as a catalyst, giving multicyclic spiro-1,3-indandiones in moderate yields with enantioselectivities as well as diastereoselectivities [39]. However, to the best of our knowledge, arylidenemalonates have not been employed as substrates in the cascade reaction of curcumins employing a biphasic solvent system for the synthesis of highly functionalized cyclohexanones and tetrahydrochromenes.
In view of the above, we report herein cascade double and triple Michael reactions of curcumins with arylidenemalonates in the presence of a base (KOH) and a phase-transfer catalyst (PTC) in a biphasic medium (toluene–H2O) at room temperature, leading to highly functionalized cyclohexanones and tetrahydrochromenones as major and minor products, respectively, in moderate to high yield and excellent diastereoselectivity.
Results and Discussion
Initially, the reaction of 1a with 2a was carried out with 3.0 equiv of aq KOH as base and toluene as a solvent in the presence of 20 mol % tetrabutylammonium bromide (TBAB) as a phase-transfer catalyst at room temperature for 24 h (Table 1, entry 1). To our delight, the formation of double Michael addition product 3a was observed in 31% yield. The reaction with 6.0 equiv of KOH resulted in an increase in the yield of 3a (72%) and also the formation of 4a in low yield (13%, Table 1, entry 2). Next, the THF–toluene solvent system was employed in the absence and presence of TBAB, affording 3a in 56%, 49% and 60% yields, respectively (Table 1, entries 3–5). As expected, no product formation was noted when the reaction was investigated without TBAB (Table 1, entry 6) and the same observation was made in the absence of base (Table 1, entry 7). Other bases such as Cs2CO3, NaOH and tetramethylguanidine (TMG) were screened in toluene as the solvent of choice (Table 1, entries 8–10), but only TMG provided the desired product 3a though in lower yield (63%, Table 1, entry 10). Interestingly, TMG furnished 55% yield of 3a in acetonitrile as solvent (Table 1, entry 11). Changing the PTC to TBAI did not have a significant effect on the yield or formation of respective products (Table 1, entry 12). Based on the optimization studies, a combination of KOH in toluene and TBAB (Table 1, entry 2) as phase-transfer catalyst was chosen as the best conditions. Although KOH could saponify the ester group, our methodology involves a biphasic reaction medium where the starting material, viz. arylidenemalonate, remains in the organic layer and prevents itself from undergoing saponification because it requires either aqueous or alcoholic (MeOH or EtOH) medium. Furthermore, deprotonation of curcumin predominates over saponification under our mild conditions (room temperature). Overall, the reaction proceeds smoothly without any unwanted side reactions such as saponification of the ester groups.
Table 1: Reaction optimisation using phase-transfer catalysts.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-177-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Base [equiv] | Catalyst [mol %] | Solvent | Time [h] | 3a Yield [%]b | 4a Yield [%]b |
| 1 | KOH (3.0) | TBAB (20) | toluene | 24 | 31 | nd |
| 2 | KOH (6.0) | TBAB (20) | toluene | 16 | 72 | 13 |
| 3 | KOH (6.0) | TBAB (20) | THF–toluene (2:8) | 48 | 56 | traces |
| 4 | KOH (6.0) | – | THF–toluene (2:8) | 48 | 49 | traces |
| 5 | KOH (6.0) | TBAB (20) | THF–toluene (8:2) | 48 | 60 | traces |
| 6c | KOH (6.0) | – | toluene | 72 | nd | nd |
| 7c | – | TBAB (20) | toluene | 72 | nd | nd |
| 8d | Cs2CO3 (6.0) | TBAB (20) | toluene | 24 | nd | nd |
| 9d | NaOH (6.0) | TBAB (20) | toluene | 48 | nd | nd |
| 10 | TMG (6.0) | TBAB (20) | toluene | 72 | 63 | nd |
| 11 | TMG (6.0) | TBAB (20) | acetonitrile | 16 | 55 | nd |
| 12 | KOH (6.0) | TBAI (20) | toluene | 72 | 69` | 11 |
aReaction scale: 1a (0.1 mmol, 1 equiv), 2a (0.12 mmol, 1.2 equiv), solvent (1.5 mL); bafter silica-gel column chromatography; cno reaction; dcomplex mixture; nd = not detected.
Having optimized the reaction conditions, we proceeded to examine the substrate scope of the developed methodology (Table 2). At first, various arylidenemalonates 2 were tested with a phenyl analog of curcumin 1. The reaction of 1a with arylidene malonates 2a–c, bearing weakly electron-withdrawing para-substituents, produced the corresponding double Michael adducts 3a–c, respectively, in good to high yields (55–75%) with excellent diastereoselectivity (Table 2, entries 1–3). At the same time, the corresponding triple Michael adducts 4a–c were also formed as minor products (12–36%). There was no reaction with ortho-nitro-substituted arylidenemalonate 2d under the optimized conditions which is attributable to the increase in electron density at the carbon β to the ester group thus inhibiting the Michael addition of curcumin (Table 2, entry 4). Arylidenemalonate 2e, bearing a weakly electron-donating substituent, reacted with 1a to afford the double Michael adduct 3e in moderate yield (46%), and the triple Michael adduct 4e was not detected (Table 2, entry 5). Bulky 1-naphthyl analog 2f delivered the double Michael adduct 3f as the only product in a very good yield (73%, Table 2, entry 6). Heteroarylidenemalonate 2g also reacted well with 1a, affording 3g in 54% yield (Table 2, entry 7). Unfortunately, no reaction occurred with aliphatic valeryl derivate 2h, presumably due to the competitive deprotonation of the active methylene group of curcumin and the allylic γ-position of the diester and/or the +I effect of the alkyl group which deactivates the β-position of the diester (Table 2, entry 8).
Table 2: Scope of alkylidenemalonates and curcumins.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-20-177-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| Entry | 1, Ar | 2, R | Time [h] | 3 | Yield [%]b | dr | 4 | Yield [%]b | dr |
| 1 | 1a, Ph | 2a, 4-ClC6H4 | 16 | 3a | 72 | >95:5 | 4a | 13 | >95:5 |
| 2 | 1a, Ph | 2b, 4-FC6H4 | 18 | 3b | 55 | >95:5 | 4b | 36 | >95:5 |
| 3 | 1a, Ph | 2c, 3-BrC6H4 | 24 | 3c | 75 | >99:5 | 4c | 12 | >95:5 |
| 4c | 1a, Ph | 2d, 2-NO2C6H4 | 36 | 3d | nd | – | 4d | nd | – |
| 5 | 1a, Ph | 2e, 4-MeC6H4 | 22 | 3e | 46 | >95:5 | 4e | nd | – |
| 6 | 1a, Ph | 2f, 1-naphthyl | 12 | 3f | 73 | >95:5 | 4f | traces | – |
| 7 | 1a, Ph | 2g, 2-furyl | 16 | 3g | 54 | >95:5 | 4g | traces | – |
| 8c | 1a, Ph | 2h, valeryl | 24 | 3h | nd | – | 4h | nd | – |
| 9 | 1b, 4-MeC6H4 | 2a, 4-ClC6H4 | 36 | 3i | 43 | >95:5 | 4i | traces | – |
| 10 | 1c, 4-BrC6H4 | 2a, 4-ClC6H4 | 48 | 3j | 35 | >95:5 | 4j | nd | – |
| 11 | 1d, 1-naphthyl | 2a, 4-ClC6H4 | 36 | 3k | 44 | >95:5 | 4k | traces | – |
| 12 | 1e, 4-MeOC6H4 | 2b, 4-FC6H4 | 18 | 3l | 54 | 79:21 | 4l | nd | – |
| 13 | 1e, 4-MeOC6H4 | 2e, 4-MeC6H4 | 48 | 3m | 42 | >95:5 | 4m | 16 | >95:5 |
| 14 | 1f, 2-Furyl | 2i, Ph | 18 | 3n | 56 | >95:5 | 4n | nd | – |
aReaction scale: 1 (0.1 mmol, 1 equiv), 2 (0.12 mmol, 1.2 equiv), TBAB (0.02 mmol, 20 mol %), toluene (1.5 mL); bafter silica-gel column chromatography; cNo reaction.; nd = not detected.
In the next set of experiments, different curcumins were examined while keeping 2a as the model arylidenemalonate (Table 2, entries 9–11). The reaction of p-tolyl and p-bromophenyl analogs of curcumin 1b,c led to the formation of double Michael adducts 3i–j in moderate yields (35–43%) over prolonged reaction time (36–48 h, Table 2, entries 9 and 10). The 1-naphthyl analog of curcumin 1d gave a moderate yield (44%) of the double Michael adduct 3k (Table 2, entry 11). The p-anisylcurcumin 1e reacted with 2b and furnished the double Michael adduct 3l in good yield (54%) but moderate diastereomeric ratio (79:21, Table 2, entry 12). Next, p-anisylcurcumin 1e when treated with p-tolylidenemalonate 2e furnished the double Michael adduct 3m in decent yield (42%) with excellent diastereoselectivity (Table 2, entry 13). Similarly, the reaction of benzylidenemalonate 2i with furylcurcumin 1f resulted in double Michael adduct 3n in 56% yield (Table 2, entry 14).
A plausible mechanism for the cascade double and triple Michael reactions is shown in Scheme 2. At first, the enolate I of curcumin 1 adds to arylidenemalonate 2 in a Michael fashion resulting enolate II. The ester enolate II might remain in equilibrium with 1,3-dicarbonyl enolate I, but the former would be trapped via cyclization involving a diastereoselective 6-endo-trig intramolecular Michael addition to the enone moiety leading to highly substituted cyclohexanone 3. The formation of triple Michael adduct 4 can be attributed to the enolate 3 undergoing yet another diastereoselective 6-endo-trig intramolecular oxa-Michael addition.
![[1860-5397-20-177-i2]](/bjoc/content/inline/1860-5397-20-177-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A plausible reaction mechanism.
Scheme 2: A plausible reaction mechanism.
The structure and stereochemistry of compounds 3 and 4 were confirmed by 1H-1H-COSY and 1H-1H-NOESY NMR experiments. This was further unambiguously established by single crystal X-ray analysis of a representative compound 4a (Figure 2). These studies confirmed that R and Ar in the cyclohexane ring of compounds 3 and 4 are trans to each other and the Ar in the dihydropyran ring of 4 is trans to R and cis to Ar in the cyclohexane ring.
![[1860-5397-20-177-2]](/bjoc/content/figures/1860-5397-20-177-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structure of 4a (CCDC 2351387).
Figure 2: X-ray structure of 4a (CCDC 2351387).
The diastereoselectivity observed in the above cascade double Michael reaction of curcumins 1 with arylidenemalonate 2 can be explained in terms of the relative stereochemistry of the substituents in the enolate arising from the first Michael addition (Figure 3). Comparison of the two possible transition states TSI and TSII for second Michael addition suggests that a severe 1,3-allylic strain destabilizes TSII where R is equatorial. Such a strain is avoided by R adopting axial orientation as in TSI, which is favoured, leading to the observed product 3. This has also been seen in previous cases where nitroalkenes and chalcones have been employed as acceptors in the reaction of curcumins [26,29].
![[1860-5397-20-177-3]](/bjoc/content/figures/1860-5397-20-177-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Origin of stereoselectivity in the double Michael addition.
Figure 3: Origin of stereoselectivity in the double Michael addition.
In order to demonstrate the synthetic utility of our methodology, we performed a scale-up reaction with representative starting materials, viz. 1a and 2b on a 1.0 mmol scale (Scheme 3). The reaction required slightly more time and resulted in the corresponding double and triple Michael adducts 3b and 4b, respectively, in marginally lower yields (see also Table 2, entry 2).
![[1860-5397-20-177-i3]](/bjoc/content/inline/1860-5397-20-177-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
A hitherto unexplored cascade double and triple Michael reactions of curcumins with arylidenemalonates is reported here. The double Michael reactions lead to highly functionalized cyclohexanones as exclusive or predominant products with excellent diastereoselectivity. On the other hand, triple Michael reactions took place in a few cases, affording highly functionalized tetrahydrochromen-4-ones as minor products with complete diastereoselectivity. The reactions were carried out in the presence of aq KOH using TBAB as a suitable phase transfer catalyst in a biphasic medium at room temperature. The scalability of the reaction has also been demonstrated. Our future efforts will involve performing an asymmetric version of this reaction using chiral phase-transfer catalysts and the results will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data. | ||
| Format: PDF | Size: 676.4 KB | Download |
| Supporting Information File 2: Copies of NMR spectra of all new compounds. | ||
| Format: PDF | Size: 2.9 MB | Download |
| Supporting Information File 3: Crystallographic information file of compound 4a. | ||
| Format: CIF | Size: 2.1 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Marco-Contelles, J.; Molina, M. T.; Anjum, S. Chem. Rev. 2004, 104, 2857–2900. doi:10.1021/cr980013j
Return to citation in text: [1] -
Imuta, S.; Ochiai, S.; Kuribayashi, M.; Chida, N. Tetrahedron Lett. 2003, 44, 5047–5051. doi:10.1016/s0040-4039(03)01186-9
Return to citation in text: [1] -
Fukuyama, Y.; Kuwayama, A.; Minami, H. Chem. Pharm. Bull. 1997, 45, 947–949. doi:10.1248/cpb.45.947
Return to citation in text: [1] -
Siegel, D. R.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 1048–1049. doi:10.1021/ja057418n
Return to citation in text: [1] -
Leung, E.; Rewcastle, G. W.; Joseph, W. R.; Rosengren, R. J.; Larsen, L.; Baguley, B. C. Invest. New Drugs 2012, 30, 2103–2112. doi:10.1007/s10637-011-9768-4
Return to citation in text: [1] -
Markaverich, B. M.; Schauweker, T. H.; Gregory, R. R.; Varma, M.; Kittrell, F. S.; Medina, D.; Varma, R. S. Cancer Res. 1992, 52, 2482–2488.
Return to citation in text: [1] -
Pulkkinen, J.; Aburel, P. S.; Halland, N.; Jørgensen, K. A. Adv. Synth. Catal. 2004, 346, 1077–1080. doi:10.1002/adsc.200404115
Return to citation in text: [1] -
He, P.; Liu, X.; Shi, J.; Lin, L.; Feng, X. Org. Lett. 2011, 13, 936–939. doi:10.1021/ol1029832
Return to citation in text: [1] -
Wei, Q.; Gong, L.-Z. Org. Lett. 2010, 12, 1008–1011. doi:10.1021/ol100020v
Return to citation in text: [1] -
Zhou, M.-Q.; Zuo, J.; Cui, B.-D.; Zhao, J.-Q.; You, Y.; Bai, M.; Chen, Y.-Z.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron 2014, 70, 5787–5793. doi:10.1016/j.tet.2014.06.042
Return to citation in text: [1] -
Esatbeyoglu, T.; Huebbe, P.; Ernst, I. M. A.; Chin, D.; Wagner, A. E.; Rimbach, G. Angew. Chem., Int. Ed. 2012, 51, 5308–5332. doi:10.1002/anie.201107724
Return to citation in text: [1] -
Liang, G. Curr. Pharm. Des. 2013, 19, 1965. doi:10.2174/1381612811319110001
Return to citation in text: [1] -
Alarcón de la Lastra, C. Mol. Nutr. Food Res. 2008, 52, 985. doi:10.1002/mnfr.200890036
Return to citation in text: [1] -
Banerjee, S.; Chakravarty, A. R. Acc. Chem. Res. 2015, 48, 2075–2083. doi:10.1021/acs.accounts.5b00127
Return to citation in text: [1] -
Vyas, A.; Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F. Curr. Pharm. Des. 2013, 19, 2047–2069. doi:10.2174/1381612811319110007
Return to citation in text: [1] -
Padhye, S.; Chavan, D.; Pandey, S.; Deshpande, J.; Swamy, K. V.; Sarkar, F. H. Mini-Rev. Med. Chem. 2010, 10, 372–387. doi:10.2174/138955710791330891
Return to citation in text: [1] -
Tomeh, M. A.; Hadianamrei, R.; Zhao, X. Int. J. Mol. Sci. 2019, 20, 1033. doi:10.3390/ijms20051033
Return to citation in text: [1] -
Anand, P.; Sundaram, C.; Jhurani, S.; Kunnumakkara, A. B.; Aggarwal, B. B. Cancer Lett. 2008, 267, 133–164. doi:10.1016/j.canlet.2008.03.025
Return to citation in text: [1] -
Izadi, M.; Sadri, N.; Abdi, A.; Zadeh, M. M. R.; jalaei, D.; Ghazimoradi, M. M.; Shouri, S.; Tahmasebi, S. GeroScience 2024, 46, 2933–2950. doi:10.1007/s11357-024-01092-5
Return to citation in text: [1] -
Mazumder, A.; Neamati, N.; Sunder, S.; Schulz, J.; Pertz, H.; Eich, E.; Pommier, Y. J. Med. Chem. 1997, 40, 3057–3063. doi:10.1021/jm970190x
Return to citation in text: [1] -
Catanzaro, M.; Corsini, E.; Rosini, M.; Racchi, M.; Lanni, C. Molecules 2018, 23, 2778. doi:10.3390/molecules23112778
Return to citation in text: [1] -
Nurfina, A. N.; Reksohadiprodjo, M. S.; Timmerman, H.; Jenie, U. A.; Sugiyanto, D.; van der Goot, H. Eur. J. Med. Chem. 1997, 32, 321–328. doi:10.1016/s0223-5234(97)89084-8
Return to citation in text: [1] -
Sharma, O. P. Biochem. Pharmacol. 1976, 25, 1811–1812. doi:10.1016/0006-2952(76)90421-4
Return to citation in text: [1] -
Abdou, M. M.; El-Saeed, R. A.; Bondock, S. Arabian J. Chem. 2019, 12, 974–1003. doi:10.1016/j.arabjc.2015.06.029
Return to citation in text: [1] -
Laha, B.; Tiwari, A. R.; Gravel, E.; Doris, E.; Namboothiri, I. N. N. Org. Biomol. Chem. 2024, 22, 1346–1359. doi:10.1039/d3ob01734f
Return to citation in text: [1] [2] -
Ayyagari, N.; Jose, D.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2011, 52, 258–262. doi:10.1016/j.tetlet.2010.11.017
Return to citation in text: [1] [2] [3] -
Namboothiri, I. N. N.; Ayyagari, N.; Jose, D. Curcumin derivatives. U.S. Patent US8410177B2, April 2, 2013.
Return to citation in text: [1] -
Ayyagari, N.; Namboothiri, I. N. N. Tetrahedron: Asymmetry 2012, 23, 605–610. doi:10.1016/j.tetasy.2012.04.011
Return to citation in text: [1] -
Ayyagari, N.; Mehta, A.; Gopi, E.; Deb, I.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron 2013, 69, 5973–5980. doi:10.1016/j.tet.2013.04.083
Return to citation in text: [1] [2] [3] -
Lal, S.; Chowdhury, A.; Namboothiri, I. N. N. Tetrahedron 2017, 73, 1297–1305. doi:10.1016/j.tet.2017.01.003
Return to citation in text: [1] -
Nair, D. K.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2012, 53, 3349–3352. doi:10.1016/j.tetlet.2012.04.084
Return to citation in text: [1] -
Bera, K.; Ayyagari, N.; Satam, N.; Namboothiri, I. N. N. Org. Biomol. Chem. 2020, 18, 140–153. doi:10.1039/c9ob01974j
Return to citation in text: [1] -
Siva Sankara, C.; Bhagat, S.; Chandra, A.; Namboothiri, I. N. N. Eur. J. Org. Chem. 2023, 26, e202300069. doi:10.1002/ejoc.202300069
Return to citation in text: [1] -
Laha, B.; Suresh, A.; Namboothiri, I. N. N. Org. Biomol. Chem. 2023, 21, 1872–1877. doi:10.1039/d2ob02211g
Return to citation in text: [1] -
Li, W.; Wu, W.; Yu, F.; Huang, H.; Liang, X.; Ye, J. Org. Biomol. Chem. 2011, 9, 2505. doi:10.1039/c0ob00757a
Return to citation in text: [1] -
Yin, X.-G.; Liu, X.-Y.; Hu, Z.-P.; Yan, M. Org. Biomol. Chem. 2012, 10, 1506. doi:10.1039/c2ob06995d
Return to citation in text: [1] -
Sahu, P. K.; Sahu, P. K.; Gupta, S. K.; Thavaselvam, D.; Agarwal, D. D. Eur. J. Med. Chem. 2012, 54, 366–378. doi:10.1016/j.ejmech.2012.05.020
Return to citation in text: [1] -
Bhuvaneswari, K.; Sivaguru, P.; Lalitha, A. ChemistrySelect 2017, 2, 11552–11560. doi:10.1002/slct.201702406
Return to citation in text: [1] -
Luo, N.-h.; Sun, X.; Wei, W.-t.; Zhang, X.-j.; Yan, M. Tetrahedron: Asymmetry 2013, 24, 402–408. doi:10.1016/j.tetasy.2013.02.014
Return to citation in text: [1]
| 39. | Luo, N.-h.; Sun, X.; Wei, W.-t.; Zhang, X.-j.; Yan, M. Tetrahedron: Asymmetry 2013, 24, 402–408. doi:10.1016/j.tetasy.2013.02.014 |
| 37. | Sahu, P. K.; Sahu, P. K.; Gupta, S. K.; Thavaselvam, D.; Agarwal, D. D. Eur. J. Med. Chem. 2012, 54, 366–378. doi:10.1016/j.ejmech.2012.05.020 |
| 38. | Bhuvaneswari, K.; Sivaguru, P.; Lalitha, A. ChemistrySelect 2017, 2, 11552–11560. doi:10.1002/slct.201702406 |
| 1. | Marco-Contelles, J.; Molina, M. T.; Anjum, S. Chem. Rev. 2004, 104, 2857–2900. doi:10.1021/cr980013j |
| 2. | Imuta, S.; Ochiai, S.; Kuribayashi, M.; Chida, N. Tetrahedron Lett. 2003, 44, 5047–5051. doi:10.1016/s0040-4039(03)01186-9 |
| 11. | Esatbeyoglu, T.; Huebbe, P.; Ernst, I. M. A.; Chin, D.; Wagner, A. E.; Rimbach, G. Angew. Chem., Int. Ed. 2012, 51, 5308–5332. doi:10.1002/anie.201107724 |
| 35. | Li, W.; Wu, W.; Yu, F.; Huang, H.; Liang, X.; Ye, J. Org. Biomol. Chem. 2011, 9, 2505. doi:10.1039/c0ob00757a |
| 7. | Pulkkinen, J.; Aburel, P. S.; Halland, N.; Jørgensen, K. A. Adv. Synth. Catal. 2004, 346, 1077–1080. doi:10.1002/adsc.200404115 |
| 8. | He, P.; Liu, X.; Shi, J.; Lin, L.; Feng, X. Org. Lett. 2011, 13, 936–939. doi:10.1021/ol1029832 |
| 9. | Wei, Q.; Gong, L.-Z. Org. Lett. 2010, 12, 1008–1011. doi:10.1021/ol100020v |
| 10. | Zhou, M.-Q.; Zuo, J.; Cui, B.-D.; Zhao, J.-Q.; You, Y.; Bai, M.; Chen, Y.-Z.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron 2014, 70, 5787–5793. doi:10.1016/j.tet.2014.06.042 |
| 36. | Yin, X.-G.; Liu, X.-Y.; Hu, Z.-P.; Yan, M. Org. Biomol. Chem. 2012, 10, 1506. doi:10.1039/c2ob06995d |
| 5. | Leung, E.; Rewcastle, G. W.; Joseph, W. R.; Rosengren, R. J.; Larsen, L.; Baguley, B. C. Invest. New Drugs 2012, 30, 2103–2112. doi:10.1007/s10637-011-9768-4 |
| 6. | Markaverich, B. M.; Schauweker, T. H.; Gregory, R. R.; Varma, M.; Kittrell, F. S.; Medina, D.; Varma, R. S. Cancer Res. 1992, 52, 2482–2488. |
| 33. | Siva Sankara, C.; Bhagat, S.; Chandra, A.; Namboothiri, I. N. N. Eur. J. Org. Chem. 2023, 26, e202300069. doi:10.1002/ejoc.202300069 |
| 34. | Laha, B.; Suresh, A.; Namboothiri, I. N. N. Org. Biomol. Chem. 2023, 21, 1872–1877. doi:10.1039/d2ob02211g |
| 3. | Fukuyama, Y.; Kuwayama, A.; Minami, H. Chem. Pharm. Bull. 1997, 45, 947–949. doi:10.1248/cpb.45.947 |
| 4. | Siegel, D. R.; Danishefsky, S. J. J. Am. Chem. Soc. 2006, 128, 1048–1049. doi:10.1021/ja057418n |
| 25. | Laha, B.; Tiwari, A. R.; Gravel, E.; Doris, E.; Namboothiri, I. N. N. Org. Biomol. Chem. 2024, 22, 1346–1359. doi:10.1039/d3ob01734f |
| 26. | Ayyagari, N.; Jose, D.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2011, 52, 258–262. doi:10.1016/j.tetlet.2010.11.017 |
| 27. | Namboothiri, I. N. N.; Ayyagari, N.; Jose, D. Curcumin derivatives. U.S. Patent US8410177B2, April 2, 2013. |
| 26. | Ayyagari, N.; Jose, D.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2011, 52, 258–262. doi:10.1016/j.tetlet.2010.11.017 |
| 30. | Lal, S.; Chowdhury, A.; Namboothiri, I. N. N. Tetrahedron 2017, 73, 1297–1305. doi:10.1016/j.tet.2017.01.003 |
| 25. | Laha, B.; Tiwari, A. R.; Gravel, E.; Doris, E.; Namboothiri, I. N. N. Org. Biomol. Chem. 2024, 22, 1346–1359. doi:10.1039/d3ob01734f |
| 29. | Ayyagari, N.; Mehta, A.; Gopi, E.; Deb, I.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron 2013, 69, 5973–5980. doi:10.1016/j.tet.2013.04.083 |
| 31. | Nair, D. K.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2012, 53, 3349–3352. doi:10.1016/j.tetlet.2012.04.084 |
| 32. | Bera, K.; Ayyagari, N.; Satam, N.; Namboothiri, I. N. N. Org. Biomol. Chem. 2020, 18, 140–153. doi:10.1039/c9ob01974j |
| 24. | Abdou, M. M.; El-Saeed, R. A.; Bondock, S. Arabian J. Chem. 2019, 12, 974–1003. doi:10.1016/j.arabjc.2015.06.029 |
| 26. | Ayyagari, N.; Jose, D.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron Lett. 2011, 52, 258–262. doi:10.1016/j.tetlet.2010.11.017 |
| 29. | Ayyagari, N.; Mehta, A.; Gopi, E.; Deb, I.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron 2013, 69, 5973–5980. doi:10.1016/j.tet.2013.04.083 |
| 12. | Liang, G. Curr. Pharm. Des. 2013, 19, 1965. doi:10.2174/1381612811319110001 |
| 13. | Alarcón de la Lastra, C. Mol. Nutr. Food Res. 2008, 52, 985. doi:10.1002/mnfr.200890036 |
| 14. | Banerjee, S.; Chakravarty, A. R. Acc. Chem. Res. 2015, 48, 2075–2083. doi:10.1021/acs.accounts.5b00127 |
| 15. | Vyas, A.; Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F. Curr. Pharm. Des. 2013, 19, 2047–2069. doi:10.2174/1381612811319110007 |
| 16. | Padhye, S.; Chavan, D.; Pandey, S.; Deshpande, J.; Swamy, K. V.; Sarkar, F. H. Mini-Rev. Med. Chem. 2010, 10, 372–387. doi:10.2174/138955710791330891 |
| 17. | Tomeh, M. A.; Hadianamrei, R.; Zhao, X. Int. J. Mol. Sci. 2019, 20, 1033. doi:10.3390/ijms20051033 |
| 18. | Anand, P.; Sundaram, C.; Jhurani, S.; Kunnumakkara, A. B.; Aggarwal, B. B. Cancer Lett. 2008, 267, 133–164. doi:10.1016/j.canlet.2008.03.025 |
| 19. | Izadi, M.; Sadri, N.; Abdi, A.; Zadeh, M. M. R.; jalaei, D.; Ghazimoradi, M. M.; Shouri, S.; Tahmasebi, S. GeroScience 2024, 46, 2933–2950. doi:10.1007/s11357-024-01092-5 |
| 20. | Mazumder, A.; Neamati, N.; Sunder, S.; Schulz, J.; Pertz, H.; Eich, E.; Pommier, Y. J. Med. Chem. 1997, 40, 3057–3063. doi:10.1021/jm970190x |
| 21. | Catanzaro, M.; Corsini, E.; Rosini, M.; Racchi, M.; Lanni, C. Molecules 2018, 23, 2778. doi:10.3390/molecules23112778 |
| 22. | Nurfina, A. N.; Reksohadiprodjo, M. S.; Timmerman, H.; Jenie, U. A.; Sugiyanto, D.; van der Goot, H. Eur. J. Med. Chem. 1997, 32, 321–328. doi:10.1016/s0223-5234(97)89084-8 |
| 23. | Sharma, O. P. Biochem. Pharmacol. 1976, 25, 1811–1812. doi:10.1016/0006-2952(76)90421-4 |
| 28. | Ayyagari, N.; Namboothiri, I. N. N. Tetrahedron: Asymmetry 2012, 23, 605–610. doi:10.1016/j.tetasy.2012.04.011 |
| 29. | Ayyagari, N.; Mehta, A.; Gopi, E.; Deb, I.; Mobin, S. M.; Namboothiri, I. N. N. Tetrahedron 2013, 69, 5973–5980. doi:10.1016/j.tet.2013.04.083 |
© 2024 Nair et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.