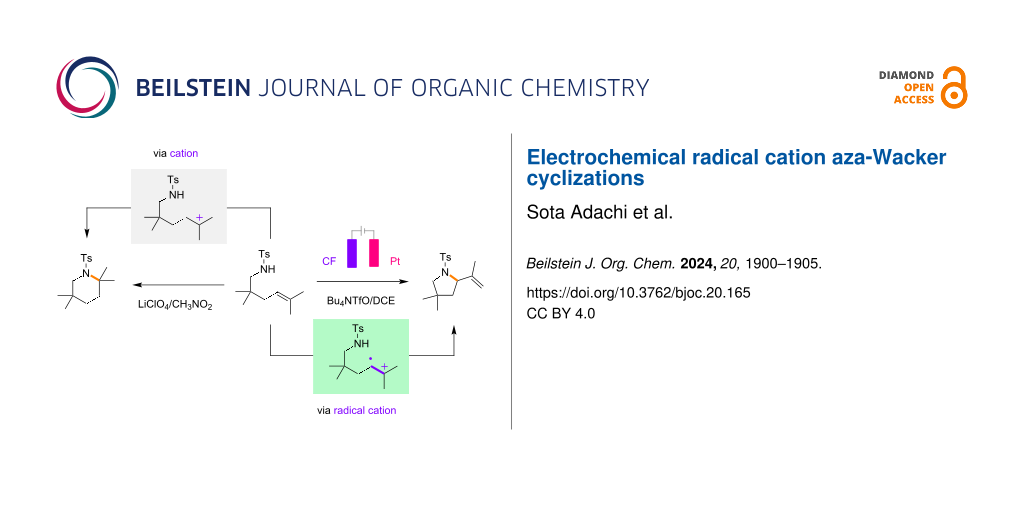
Abstract
Electrochemical or photochemical single-electron oxidation of bench-stable substrates can generate radical cations that offer unique reactivities as intermediates in various bond-formation processes. Such intermediates can potentially take part in both radical and ionic bond formation; however, the mechanisms involved are complicated and not fully understood. Herein, we report electrochemical radical cation aza-Wacker cyclizations under acidic conditions, which are expected to proceed via radical cations generated by single-electron oxidation of alkenes.

Graphical Abstract
Introduction
Activating bench-stable substrates is the first step to driving bond formation and/or cleavage. Therefore, the discovery of new modes for activation leads to reaction advancements. Electrochemical [1-5] and photochemical [6-10] reactions that induce single-electron reduction and oxidation are widely used in modern synthetic organic chemistry [11-15]. Single-electron oxidation of bench-stable substrates can generate radical cations that offer unique reactivities as intermediates for various bond-formation processes (also true for reduction). Because the reactivities of radicals and ions are fundamentally different, their creative use may pave the way for complementary bond formation. This merging is unique and such intermediates could potentially take part in both radical and ionic bond formation. However, the mechanisms involved can be complicated and are not fully understood.
Alkenes and styrenes are representative radical cation precursors that are widely used to realize the formation of unique bonds. The respective radical cations are trapped by various nucleophiles under radical and/or ion control, where kinetic and/or thermodynamic effects are expected to be dominant. Typical examples that clearly show the difference in such reactivities are intramolecular cyclizations (Scheme 1). A radical cyclization generates a five-membered ring with a less-stable primary radical, while a six-membered ring with a secondary cation is obtained through ionic cyclization. When such intramolecular cyclizations are expected to proceed via radical cations, there are several interpretations of the mechanisms involved, since radical and ionic cyclizations are both possible.
![[1860-5397-20-165-i1]](/bjoc/content/inline/1860-5397-20-165-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Radical and ionic intramolecular cyclizations.
Scheme 1: Radical and ionic intramolecular cyclizations.
In this context, electrochemical and photochemical aza-Wacker cyclizations have provided interesting models for mechanistic discussion (Scheme 2). For example, Moeller reported electrochemical reactions under basic conditions, which were proposed to proceed via radicals [16-18]. Xu also reported electrochemical reactions via radicals, which were generated through proton-coupled electron transfer [19]. On the other hand, Yoon reported photochemical reactions under acidic conditions, which were proposed to proceed via radical cations [20]. Since electrochemical and photochemical aza-Wacker cyclizations can offer ring systems that are difficult to construct through state-of-the-art palladium-catalyzed methods, the mechanistic understanding of these cyclizations would be of great help to expand their synthetic utility. Described herein are electrochemical aza-Wacker cyclizations under acidic conditions, which are expected to proceed via radical cations.
![[1860-5397-20-165-i2]](/bjoc/content/inline/1860-5397-20-165-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical and photochemical aza-Wacker cyclizations.
Scheme 2: Electrochemical and photochemical aza-Wacker cyclizations.
Results and Discussion
The present work began by examining the electrochemical aza-Wacker cyclization using the alkene 1 as a model (Table 1). Based on the conditions reported by Yoon and Moeller, the initial screening was carried out using tetrabutylammonium triflate (Bu4NOTf)/1,2-dichloroethane (1,2-DCE) solution. Carbon felt (CF) was used as an anode instead of reticulated vitreous carbon (RVC), with platinum (Pt) as a cathode. To our delight, a constant-current condition at 1 mA was productive, and the desired five-membered pyrrolidine 2 was obtained in high yield (Table 1, entry 2). During the screening of conditions, the addition of acetonitrile (CH3CN) was found to be effective, probably due to the increased conductivity of the electrolyte solution (Table 1, entry 1). The reaction did not take place without electricity and most of the starting material was recovered (Table 1, entry 3). The addition of trifluoroacetic acid (TFA) was advantageous in terms of the reproducibility, which was in good accordance with the observation reported by Yoon (Table 1, entry 4). The use of acetic acid (AcOH) instead of TFA gave a slightly lower yield of the five-membered pyrrolidine 2 (Table 1, entry 5). Although a constant-potential condition at 1.8 V was also productive, the constant-current condition gave better results (Table 1, entry 6). Previously, we reported that lithium perchlorate (LiClO4)/nitromethane (CH3NO2) solution was an effective medium to facilitate radical cation reactions [21-25]. However, interestingly, it was not productive for the electrochemical aza-Wacker cyclization (Table 1, entry 7) and the six-membered piperidine 3, instead of the five-membered pyrrolidine 2, was obtained in excellent yield without electricity (Table 1, entry 8). Thus, it is proposed that the electrochemical aza-Wacker cyclization under acidic conditions proceeded via radical cations to give five-membered pyrrolidine 2, while the six-membered piperidine 3 is formed through ionic cyclization under non-electrochemical conditions.
Table 1: Control studies for electrochemical aza-Wacker cyclization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-165-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviation from the optimal condition | Yields of 2 + 3b |
| 1 | 97 (0) + 0 | |
| 2 | no CH3CN | 82 (0) + 0 |
| 3 | no current | 0 (75) + 0 |
| 4 | no TFA | 81 (3) + 0 |
| 5 | AcOH instead of TFA | 66 (5) + 0 |
| 6 | constant potential at 1.8 V | 74 (0) + 0 |
| 7c | LiClO4/CH3NO2 | 0 (0) + 0 |
| 8c | LiClO4/CH3NO2, no current | 0 (0) + 96 |
aReaction conditions: alkene 1 (0.20 mmol), Bu4NOTf (0.1 M), TFA (1 equiv), CH3CN (0.4 mL), and 1,2-DCE (3.6 mL). bDetermined by NMR analysis. The recovered starting material is reported in parentheses. cLiClO4 (1 M) instead of Bu4NOTf (0.1 M).
With the optimized conditions in hand, the scope of the electrochemical aza-Wacker cyclization was investigated (Scheme 3). Various aryl sulfonamides 4–6 were compatible to give the respective five-membered pyrrolidines, except for that possessing a 2-nitro group 7. As discussed later with cyclic voltammetric studies, the electron density in the aryl rings does not seem to have a significant impact on the reaction. While benzyl sulfonamide 8 was productive under the optimized conditions, unprotected amine 9 was not compatible. Although gem-dimethyl groups installed at the tether should have a positive impact on intramolecular cyclization, they were not essential for the reaction (10).
![[1860-5397-20-165-i3]](/bjoc/content/inline/1860-5397-20-165-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of electrochemical aza-Wacker cyclization. Reaction conditions: the alkene (0.20 mmol), Bu4NOTf (0.1 M), TFA (1 equiv), CH3CN (0.4 mL), and 1,2-DCE (3.6 mL). Yields reported here are isolated yields.
Scheme 3: Scope of electrochemical aza-Wacker cyclization. Reaction conditions: the alkene (0.20 mmol), Bu4NO...
In order to obtain mechanistic insight into the aza-Wacker cyclization, differently substituted alkenes 11, 14 were prepared and subjected to the reaction under electrochemical and non-electrochemical conditions (Scheme 4). In the case of the trisubstituted alkene 11, the six-membered anti-Markovnikov product 12 was selectively obtained under electrochemical conditions, while the five-membered Markovnikov product 13 was obtained in good yield under non-electrochemical conditions. In the case of the tetrasubstituted alkene 14, the five-membered pyrrolidine 15 was selectively obtained under electrochemical conditions, while both the five-membered pyrrolidine 16 and six-membered piperidine 17 were obtained in good mass balance under non-electrochemical conditions. Although the detailed mechanism remains an open question, the electrochemical aza-Wacker cyclizations might be radical reactions rather than ionic ones, since the six-membered piperidine was not obtained from the tetrasubstituted alkene 14.
![[1860-5397-20-165-i4]](/bjoc/content/inline/1860-5397-20-165-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanistic studies of aza-Wacker cyclization. A: Electrochemical (Bu4NOTf in CH3CN/1,2-DCE), B: non-electrochemical (LiClO4 in CH3NO2).
Scheme 4: Mechanistic studies of aza-Wacker cyclization. A: Electrochemical (Bu4NOTf in CH3CN/1,2-DCE), B: no...
Cyclic voltammetric studies have provided further mechanistic insights into electrochemical aza-Wacker cyclizations. As reported by Yoon, a trisubstituted alkene is oxidized at significantly lower potential than aryl sulfonamides, suggesting that the reactions were initiated by single-electron oxidation of the alkenes. Although a drop in oxidation potential for the alkene was observed when tethered to an aryl sulfonamide, as detailed by Moeller, rapid intramolecular cyclization would be the key [26-28]. We also measured cyclic voltammograms for aryl sulfonamides with and without trisubstituted alkenes (Figure 1). As described above, the electron density in the aryl rings does not seem to have a significant impact on the reaction, since alkenes possessing methyl 2, methoxy 5, and trifluoromethyl 6 groups were all high yielding. This observation was supported by the cyclic voltammetric studies, namely, their oxidation potentials were similar. This suggests that the reactions are initiated by single-electron oxidation of alkenes instead of aryl sulfonamides, leading to unique radical cation aza-Wacker cyclizations. The cyclic voltammogram of the aryl sulfonamide without a trisubstituted alkene provides clear-cut experimental evidence of this, since the oxidation potential was recorded at a much higher value.
![[1860-5397-20-165-1]](/bjoc/content/figures/1860-5397-20-165-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Cyclic voltammograms for aryl sulfonamides.
Figure 1: Cyclic voltammograms for aryl sulfonamides.
Conclusion
In conclusion, we have demonstrated that electrochemical aza-Wacker cyclizations are enabled under acidic conditions, and are expected to proceed via radical cations. Synthetic outcomes and cyclic voltammetric studies suggest that the reactions are initiated by single-electron oxidation of the alkenes instead of the aryl sulfonamides. Although the detailed mechanism remains an open question, the electrochemical radical cation aza-Wacker cyclizations might be radical reactions rather than ionic ones, since five-membered pyrrolidine formation is preferred over six-membered piperidine formation. Further mechanistic studies of the electrochemical radical cation aza-Wacker cyclizations are underway in our laboratory.
Experimental
Electrochemical aza-Wacker cyclizations: The appropriate alkene (0.20 mmol), TFA (0.20 mmol), and CH3CN (0.4 mL) were added to a solution of Bu4NOTf/1,2-DCE (0.10 M, 3.6 mL) while stirring at room temperature. The resulting reaction mixture was electrolyzed at 1 mA using a CF anode (10 mm × 10 mm) and a Pt cathode (10 mm × 20 mm) in an undivided cell with stirring. The solution was diluted with water and extracted with dichloromethane. The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Yields were determined by 1H NMR analysis using benzaldehyde as an internal standard (Table 1). Silica gel column chromatography (hexane/ethyl acetate) gave the corresponding ring compounds.
Supporting Information
| Supporting Information File 1: General remarks and characterization data, including copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 4.5 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Chan, A. Y.; Perry, I. B.; Bissonnette, N. B.; Buksh, B. F.; Edwards, G. A.; Frye, L. I.; Garry, O. L.; Lavagnino, M. N.; Li, B. X.; Liang, Y.; Mao, E.; Millet, A.; Oakley, J. V.; Reed, N. L.; Sakai, H. A.; Seath, C. P.; MacMillan, D. W. C. Chem. Rev. 2022, 122, 1485–1542. doi:10.1021/acs.chemrev.1c00383
Return to citation in text: [1] -
Genzink, M. J.; Kidd, J. B.; Swords, W. B.; Yoon, T. P. Chem. Rev. 2022, 122, 1654–1716. doi:10.1021/acs.chemrev.1c00467
Return to citation in text: [1] -
Holmberg-Douglas, N.; Nicewicz, D. A. Chem. Rev. 2022, 122, 1925–2016. doi:10.1021/acs.chemrev.1c00311
Return to citation in text: [1] -
Allen, A. R.; Noten, E. A.; Stephenson, C. R. J. Chem. Rev. 2022, 122, 2695–2751. doi:10.1021/acs.chemrev.1c00388
Return to citation in text: [1] -
Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, 0052. doi:10.1038/s41570-017-0052
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Kärkäs, M. D. Chem. Soc. Rev. 2018, 47, 5786–5865. doi:10.1039/c7cs00619e
Return to citation in text: [1] -
Malapit, C. A.; Prater, M. B.; Cabrera-Pardo, J. R.; Li, M.; Pham, T. D.; McFadden, T. P.; Blank, S.; Minteer, S. D. Chem. Rev. 2022, 122, 3180–3218. doi:10.1021/acs.chemrev.1c00614
Return to citation in text: [1] -
Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060
Return to citation in text: [1] -
Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. doi:10.1002/anie.201712732
Return to citation in text: [1] -
Huang, H.; Steiniger, K. A.; Lambert, T. H. J. Am. Chem. Soc. 2022, 144, 12567–12583. doi:10.1021/jacs.2c01914
Return to citation in text: [1] -
Murray, P. R. D.; Cox, J. H.; Chiappini, N. D.; Roos, C. B.; McLoughlin, E. A.; Hejna, B. G.; Nguyen, S. T.; Ripberger, H. H.; Ganley, J. M.; Tsui, E.; Shin, N. Y.; Koronkiewicz, B.; Qiu, G.; Knowles, R. R. Chem. Rev. 2022, 122, 2017–2291. doi:10.1021/acs.chemrev.1c00374
Return to citation in text: [1] -
Tay, N. E. S.; Lehnherr, D.; Rovis, T. Chem. Rev. 2022, 122, 2487–2649. doi:10.1021/acs.chemrev.1c00384
Return to citation in text: [1] -
Liu, J.; Lu, L.; Wood, D.; Lin, S. ACS Cent. Sci. 2020, 6, 1317–1340. doi:10.1021/acscentsci.0c00549
Return to citation in text: [1] -
Barham, J. P.; König, B. Angew. Chem., Int. Ed. 2020, 59, 11732–11747. doi:10.1002/anie.201913767
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2008, 130, 13542–13543. doi:10.1021/ja806259z
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2010, 132, 2839–2844. doi:10.1021/ja910586v
Return to citation in text: [1] -
Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j
Return to citation in text: [1] -
Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835
Return to citation in text: [1] -
Reed, N. L.; Lutovsky, G. A.; Yoon, T. P. J. Am. Chem. Soc. 2021, 143, 6065–6070. doi:10.1021/jacs.1c02747
Return to citation in text: [1] -
Okada, Y.; Chiba, K. Chem. Rev. 2018, 118, 4592–4630. doi:10.1021/acs.chemrev.7b00400
Return to citation in text: [1] -
Okada, Y. Electrochemistry 2020, 88, 497–506. doi:10.5796/electrochemistry.20-00088
Return to citation in text: [1] -
Okada, Y. Chem. Rec. 2021, 21, 2223–2238. doi:10.1002/tcr.202100029
Return to citation in text: [1] -
Shida, N.; Imada, Y.; Okada, Y.; Chiba, K. Eur. J. Org. Chem. 2020, 570–574. doi:10.1002/ejoc.201901576
Return to citation in text: [1] -
Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/c7cc00664k
Return to citation in text: [1] -
Moeller, K. D.; Tinao, L. V. J. Am. Chem. Soc. 1992, 114, 1033–1041. doi:10.1021/ja00029a036
Return to citation in text: [1] -
Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924
Return to citation in text: [1] -
Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656
Return to citation in text: [1]
| 1. | Chan, A. Y.; Perry, I. B.; Bissonnette, N. B.; Buksh, B. F.; Edwards, G. A.; Frye, L. I.; Garry, O. L.; Lavagnino, M. N.; Li, B. X.; Liang, Y.; Mao, E.; Millet, A.; Oakley, J. V.; Reed, N. L.; Sakai, H. A.; Seath, C. P.; MacMillan, D. W. C. Chem. Rev. 2022, 122, 1485–1542. doi:10.1021/acs.chemrev.1c00383 |
| 2. | Genzink, M. J.; Kidd, J. B.; Swords, W. B.; Yoon, T. P. Chem. Rev. 2022, 122, 1654–1716. doi:10.1021/acs.chemrev.1c00467 |
| 3. | Holmberg-Douglas, N.; Nicewicz, D. A. Chem. Rev. 2022, 122, 1925–2016. doi:10.1021/acs.chemrev.1c00311 |
| 4. | Allen, A. R.; Noten, E. A.; Stephenson, C. R. J. Chem. Rev. 2022, 122, 2695–2751. doi:10.1021/acs.chemrev.1c00388 |
| 5. | Twilton, J.; Le, C.; Zhang, P.; Shaw, M. H.; Evans, R. W.; MacMillan, D. W. C. Nat. Rev. Chem. 2017, 1, 0052. doi:10.1038/s41570-017-0052 |
| 19. | Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835 |
| 16. | Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2008, 130, 13542–13543. doi:10.1021/ja806259z |
| 17. | Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2010, 132, 2839–2844. doi:10.1021/ja910586v |
| 18. | Campbell, J. M.; Xu, H.-C.; Moeller, K. D. J. Am. Chem. Soc. 2012, 134, 18338–18344. doi:10.1021/ja307046j |
| 11. | Huang, H.; Steiniger, K. A.; Lambert, T. H. J. Am. Chem. Soc. 2022, 144, 12567–12583. doi:10.1021/jacs.2c01914 |
| 12. | Murray, P. R. D.; Cox, J. H.; Chiappini, N. D.; Roos, C. B.; McLoughlin, E. A.; Hejna, B. G.; Nguyen, S. T.; Ripberger, H. H.; Ganley, J. M.; Tsui, E.; Shin, N. Y.; Koronkiewicz, B.; Qiu, G.; Knowles, R. R. Chem. Rev. 2022, 122, 2017–2291. doi:10.1021/acs.chemrev.1c00374 |
| 13. | Tay, N. E. S.; Lehnherr, D.; Rovis, T. Chem. Rev. 2022, 122, 2487–2649. doi:10.1021/acs.chemrev.1c00384 |
| 14. | Liu, J.; Lu, L.; Wood, D.; Lin, S. ACS Cent. Sci. 2020, 6, 1317–1340. doi:10.1021/acscentsci.0c00549 |
| 15. | Barham, J. P.; König, B. Angew. Chem., Int. Ed. 2020, 59, 11732–11747. doi:10.1002/anie.201913767 |
| 6. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 7. | Kärkäs, M. D. Chem. Soc. Rev. 2018, 47, 5786–5865. doi:10.1039/c7cs00619e |
| 8. | Malapit, C. A.; Prater, M. B.; Cabrera-Pardo, J. R.; Li, M.; Pham, T. D.; McFadden, T. P.; Blank, S.; Minteer, S. D. Chem. Rev. 2022, 122, 3180–3218. doi:10.1021/acs.chemrev.1c00614 |
| 9. | Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060 |
| 10. | Möhle, S.; Zirbes, M.; Rodrigo, E.; Gieshoff, T.; Wiebe, A.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. doi:10.1002/anie.201712732 |
| 26. | Moeller, K. D.; Tinao, L. V. J. Am. Chem. Soc. 1992, 114, 1033–1041. doi:10.1021/ja00029a036 |
| 27. | Xu, H.-C.; Moeller, K. D. Angew. Chem., Int. Ed. 2010, 49, 8004–8007. doi:10.1002/anie.201003924 |
| 28. | Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656 |
| 21. | Okada, Y.; Chiba, K. Chem. Rev. 2018, 118, 4592–4630. doi:10.1021/acs.chemrev.7b00400 |
| 22. | Okada, Y. Electrochemistry 2020, 88, 497–506. doi:10.5796/electrochemistry.20-00088 |
| 23. | Okada, Y. Chem. Rec. 2021, 21, 2223–2238. doi:10.1002/tcr.202100029 |
| 24. | Shida, N.; Imada, Y.; Okada, Y.; Chiba, K. Eur. J. Org. Chem. 2020, 570–574. doi:10.1002/ejoc.201901576 |
| 25. | Imada, Y.; Yamaguchi, Y.; Shida, N.; Okada, Y.; Chiba, K. Chem. Commun. 2017, 53, 3960–3963. doi:10.1039/c7cc00664k |
| 20. | Reed, N. L.; Lutovsky, G. A.; Yoon, T. P. J. Am. Chem. Soc. 2021, 143, 6065–6070. doi:10.1021/jacs.1c02747 |
© 2024 Adachi and Okada; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.