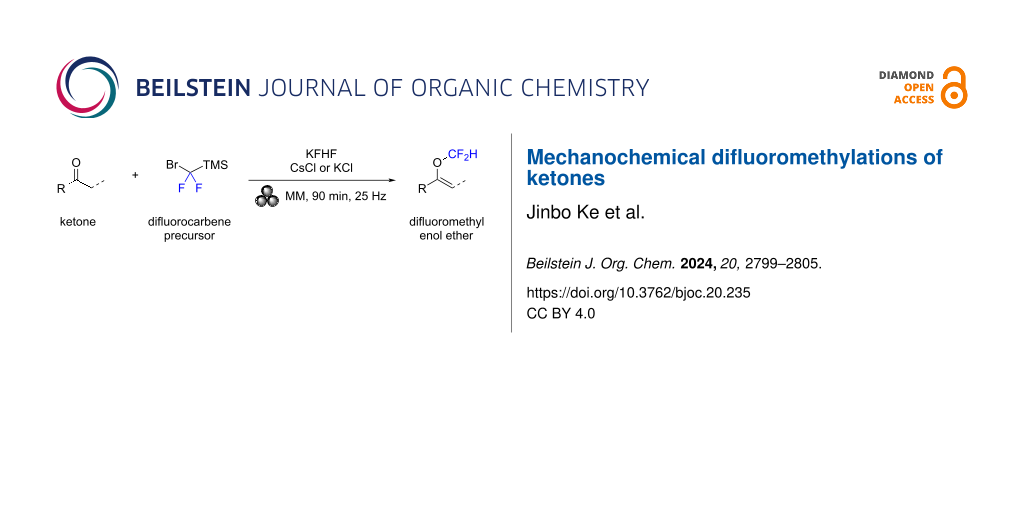
Abstract
We present a mechanochemical synthesis of difluoromethyl enol ethers. Utilizing an in situ generation of difluorocarbenes, ketones are efficiently converted to the target products under solvent-free conditions. The reactions proceed at room temperature and are complete within 90 minutes, demonstrating both efficiency and experimental simplicity.

Graphical Abstract
Introduction
In recent years, mechanochemical organic synthesis has been advanced significantly, prompting organic chemists to reconsider the necessity of solvents in their reactions [1-11]. Eliminating hazardous solvents substantially reduces the ecological footprint of organic reactions [12,13]. Beyond environmental benefits and enhanced human safety, mechanochemical reactions often feature shorter reaction times, eliminate the need for external heating, and offer alternative product selectivity [3,14]. In general, such reactions are characterized by the absorption of mechanical energy and they are influenced by several factors, including the lack of solvation, changes in morphology and rheology of the reaction mixtures during the milling, and variations in concentration and dielectric environment. Consequently, an increased reactivity can be achieved through the formation of novel reactive intermediates [15-18].
Fluorine-containing functional groups are essential structural motifs in the development of new bioactive compounds and functional materials. Compared to their non-fluorinated analogs, the presence of fluorine atoms in molecular structures can improve physicochemical and biological properties [19-27]. Among these groups, the difluoromethyl moiety has gained considerable attention [28-30]. Commonly, it is synthesized by the reaction of a nucleophile with difluorocarbene. However, the generation of difluorocarbene typically requires harsh conditions and involves toxic precursors, alongside with the risk of dimerization to tetrafluoroethylene [31]. Although this dimerization can be mitigated by controlling the concentration and reaction environment, as longer half-lives are observed for difluorocarbenes in the gas phase than in solution [32,33], it has remained a challenge to control such reactions.
Our group has recently reported a mechanochemical difluoromethylation of primary, secondary, and tertiary alcohols [34], yielding products with difluoromethoxy groups, which are promising organofluorine compounds [35-38]. Notably, also sterically hindered alcohols, which are typically less reactive in solution, could be applied under solvent-free conditions in a ball mill [39], which was attributed to a better accessibility of the difluorocarbene in the mechanochemical environment [40].
Motivated by these findings, we now explored difluoromethylation reactions with compounds bearing less nucleophilic functional groups. Ketones caught our particular attention as they contain a weak nucleophilic carbonyl oxygen adjacent to an electrophilic carbonyl carbon. Previous studies have focused on reactions of difluorocarbene with cyclic and acyclic 1,3-diones (Scheme 1A) [41-45]. Typically, they were conducted with a base to form the corresponding enolate anions which then reacted with difluorocarbene to yield difluoromethyl enol ethers. Those products are of interest because they contain a unique structural motif with potential for further functionalizations into highly diverse secondary or tertiary difluoroalkyl ethers. Dolbier and co-workers investigated reactions of difluorocarbene generated from its precursor trimethylsilyl 2,2-difluoro-2-(fluorosulfonyl)acetate (TFDA) and sodium fluoride catalyst, with simple ketones, which resulted in the formation of difluoromethyl 2,2-difluorocyclopropyl ethers (Scheme 1B). Although the reactions worked well, it is also noteworthy that the use of TFDA as reagent, liberated fluoro(trimethyl)silane (TMSF), carbon dioxide, and ozone-depleting sulfur dioxide as side products [46,47]. Later, Ichikawa and co-workers established the release of difluorocarbene from TFDA with catalytic amounts of an N-heterocyclic carbene and a base (Scheme 1C) [29,48,49]. In these reactions, difluoromethyl enol ethers were obtained, which were subsequently oxidized to yield the corresponding aryl difluoromethyl ethers. Noteworthy, however, the latter study focused mostly on cyclic ketones, with only one reported example of a difluoromethylation reaction of an acyclic substrate.
![[1860-5397-20-235-i1]](/bjoc/content/inline/1860-5397-20-235-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Overview over difluoromethyl enol ether syntheses from acyclic and cyclic 1,3-diones (A), acyclic ketones (B), and cyclic ketones (C).
Scheme 1: Overview over difluoromethyl enol ether syntheses from acyclic and cyclic 1,3-diones (A), acyclic k...
Against this background and seeing new synthetic opportunities, we wondered about reactions of mechanochemically generated difluorocarbene with simple acyclic ketones. The results and observations of this study are summarized here.
Results and Discussion
For the optimization of the reaction conditions, 4-methylacetophenone (1a) was chosen as model substrate. Under standard reaction conditions with difluorocarbene precursor TMSCF2Br (2, 2.0 equiv), activator KFHF (4.0 equiv), and grinding auxiliary CsCl (4.0 equiv), difluoromethyl enol ether 3a was obtained after 90 min reaction time at 25 Hz in 74% yield, determined by quantitative 1H NMR spectroscopy (Table 1, entry 1). The reaction was conducted in a PTFE milling equipment with two milling balls (diameter: 10 mm). Changing to a heavier milling ball (diameter: 15 mm) resulted in a yield of 67% of 3a (Table 1, entry 2). Stopping the reaction after 60 min gave product 3a in 72% yield (Table 1, entry 3). At a reaction time of 60 min, both reducing and increasing the amount of 2 (from initially used 2.0 equiv to 1.5 equiv and 3.0 equiv, respectively) reduced the yield of 3a by about 10% (Table 1, entries 3–5). Probably, with less carbene precursor the amount of generated difluorocarbene was insufficient, and with too much of it, side reactions occurred [31-33]. Next, various grinding auxiliaries were investigated at a reaction time of 60 min (Table 1, entries 6–9). A similar yield of 3a (69%) was obtained with KCl or KBr instead of CsCl (Table 1, entries 6 and 7 versus entry 3). Using NaCl, gave 3a in 64% yield (Table 1, entry 8). Finally, CsCl was substituted by silica, which, to our surprise, blocked the product formation completely (Table 1, entry 9). Apparently, the presence of an alkali halide salt was beneficial, most likely by stabilizing the consistency of the reaction mixture leading to a sufficient mixing. Silica could not fulfill this role. Lastly, water, 1,4-dioxane, chloroform, and toluene were tested in a liquid-assisted grinding (LAG) protocol (Table 1, entries 10–13). The lowest yields were obtained with water and 1,4-dioxane, providing 3a in yields of 37% and 43%, respectively (Table 1, entries 10 and 11). With the less polar solvents chloroform and toluene 3a was obtained in 62% and 68% yield, respectively (Table 1, entries 12 and 13).
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-235-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviation from the reaction conditions | Yield of 3a (%)b |
| 1 | none | 74c |
| 2 | with one PTFE milling ball (diameter: 15 mm) | 67 |
| 3 | 60 min | 72 |
| 4 | 60 min, 2 (1.5 equiv) | 61 |
| 5 | 60 min, 2 (3.0 equiv) | 60 |
| 6 | 60 min, KCl instead of CsCl | 69 |
| 7 | 60 min, KBr instead of CsCl | 69 |
| 8 | 60 min, NaCl instead of CsCl | 64 |
| 9 | 60 min, SiO2 instead of CsCl | 0 |
| 10 | LAG (H2O) | 37 |
| 11 | LAG (1,4-dioxane) | 43 |
| 12 | LAG (CHCl3) | 62 |
| 13 | LAG (toluene) | 68 |
aReaction performed with two PTFE milling balls (diameter: 10 mm) in a PTFE jar (volume: 25 mL). Liquid-assisted grinding (LAG): 0.25 µL·mg−1. bDetermined by 1H NMR spectroscopy using 1,2-dichloroethane as the internal standard. cRepetition of the experiment gave consistent results.
For comparison, the difluoromethylation of ketone 1a with difluorocarbene precursor TMSCF2Br (2) was investigated in solution (Scheme 2). The reaction conditions were chosen based on those reported by Ni, Hu and co-workers for the difluoromethylation of alcohols in solution [39]. The two activators KFHF and KOAc were investigated in a dichloromethane/water mixture at room temperature for 10 h. In both cases, the yield of 3a was negligible (with KFHF: 3%, with KOAc: nil). Following these initial attempts, the mechanochemical approach appears to be superior. However, it should also be noted that the reaction with difluorocarbene precursor 2 was not further optimized in solution.
![[1860-5397-20-235-i2]](/bjoc/content/inline/1860-5397-20-235-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Attempted difluoromethylation of 1a in solution. The reactions were performed on a 0.2 mmol scale. Method A: 2 (2.0 equiv), KFHF (4.0 equiv), CH2Cl2 (0.2 mL), H2O (0.2 mL), rt, 10 h; Method B: 2 (2.0 equiv), KOAc (4.0 equiv), CH2Cl2 (0.2 mL), H2O (0.2 mL), rt, 10 h. The yields were determined by 19F NMR spectroscopy using trifluoromethoxybenzene as the internal standard.
Scheme 2: Attempted difluoromethylation of 1a in solution. The reactions were performed on a 0.2 mmol scale. ...
Next, various ketones were investigated under the optimized reaction conditions with difluorocarbene precursor 2, KFHF (4 equiv) as activator, and CsCl or KCl (4 equiv) as grinding auxiliaries in a PTFE milling jar for 90 min at 25 Hz (Scheme 3). To get an initial efficiency estimate, the crude reaction mixtures were first analyzed by quantitative 1H NMR spectroscopy with 1,2-dichloroethane as the internal standard. After these analyses, isolating the products by column chromatography was attempted. Unfortunately, many products were highly volatile and very non-polar, rendering the purification difficult. As a result, in several cases only little or no product was obtained. Furthermore, most isolated products had only purities of ca. 90% still containing inseparable impurities (as revealed by NMR spectroscopy).
![[1860-5397-20-235-i3]](/bjoc/content/inline/1860-5397-20-235-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of ketones. The yields were determined by 1H NMR spectroscopy using 1,2-dichloroethane as the internal standard. In parentheses: yields after column chromatography (with product purities of ca. 90%). aWith CsCl. bWith KCl.
Scheme 3: Scope of ketones. The yields were determined by 1H NMR spectroscopy using 1,2-dichloroethane as the...
In the first series of substrates, acetophenone derivatives with various para-substituents were applied. Similar to methyl tolyl ketone (1a), which afforded product 3a in 74% yield, acetophenone (1b) gave 3b in 56% yield. Substrates 1c and 1d bearing a chloro or a bromo group in para position of the aryl moiety, gave the corresponding products in yields of 53% (for 3c) and 42% (for 3d). These two difluoromethyl enol ethers were also isolated by column chromatography, which afforded the products in 53% and 39% yield, respectively. Product 3e with an isobutyl group in para-position was obtained in 63% yield and isolated in 35% yield. Changing the position of the methyl group to the ortho-position led to a decrease in yield (3f: 34%). ortho-Methoxy-substituted ketone 1g provided the corresponding product 3g in 56% yield. Difluoromethyl enol ether 3h with three methyl groups was obtained in 56% yield and column chromatography allowed to isolate it in 42% yield. 2-Acetonaphthone was successfully converted to 3i in 66% yield. Besides aryl ketones, arylalkyl ketones reacted well too. Accordingly, 3j was obtained in 42% yield. Enone 1k gave 3k in 51% yield, and after isolation by column chromatography the product was obtained in 13% yield. Difluoromethyl enol ether 3l was formed from diketone 1l in 25% yield. Finally, conversions of the two cyclic ketones 1m and 1n were studied. Both gave the expected products in yields of 50% (for 3m) and 44% (for 3n).
Besides these successful transformations several ketones proved unsuitable (Scheme S1 in Supporting Information File 1). Additionally, attempted [4 + 1]-type cycloadditions of three 1-arylprop-2-en-1-ones as heteroconjugated alkenes with difluorocarbene to give 2,2-difluoro-2,3-dihydrofurans [50] remained unsuccessful (Scheme S2 in Supporting Information File 1).
Two mechanisms have been proposed for the difluoromethylation of ketones, as illustrated in Scheme 4A. In both cases, the process begins with the generation of difluorocarbene from TMSCF2Br and KFHF. This is followed by a nucleophilic attack of the oxygen atom of ketone 1 on the difluorocarbene. Subsequently, a protonation–deprotonation sequence occurs, which can either be intermolecular, involving a molecule of HF, or intramolecular, proceeding through a five-membered transition state.
![[1860-5397-20-235-i4]](/bjoc/content/inline/1860-5397-20-235-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism (A) and mechanistic investigations (B and C). The yields were determined by 1H NMR spectroscopy using 1,2-dichloroethane as internal standard.
Scheme 4: Proposed mechanism (A) and mechanistic investigations (B and C). The yields were determined by 1H N...
To clarify the mechanism, two experiments were conducted. In the first one, 2-acetonaphthone with a trideuteromethyl group (1i-d3) was subjected to the standard reaction conditions. Two products were obtained: First, 3i-d2 containing a CF2H group, and second 3i-d3 bearing a CF2D group. The yields were 57% and 15%, respectively. In the second experiment, the potential for proton exchange in difluoromethyl enol ether 3h was investigated. The compound was milled with the activator KFHF, CsCl as grinding auxiliary, and D2O in a liquid-assisted grinding process. As a result, no H/D-exchange was detected. The experimental results of both experiments suggest that the reaction predominantly proceeds through an intermolecular pathway. The occurrence of the CF2D product may be attributed to a minor intramolecular reaction path or the involvement of DF formed during the reaction.
Conclusion
In conclusion, we discovered a mechanochemical synthesis of difluoromethyl enol ethers. The products were obtained from the corresponding ketones at room temperature after a reaction time of 90 minutes. The investigation of the reaction scope revealed challenges in isolating the low-boiling non-polar products. Mechanistic studies suggested that in situ-generated difluorocarbene reacts with the ketone oxygen, followed by intermolecular protonation/deprotonation. Although the process has still synthetic limitations, also acyclic ketones can now be converted into difluoromethyl enol ethers, which have the potential for further functionalization.
Supporting Information
| Supporting Information File 1: Experimental procedures, optimization studies, compound characterization data, NMR spectra, and mechanistic investigations. | ||
| Format: PDF | Size: 3.8 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c
Return to citation in text: [1] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a
Return to citation in text: [1] -
Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a
Return to citation in text: [1] [2] -
Andersen, J.; Mack, J. Green Chem. 2018, 20, 1435–1443. doi:10.1039/c7gc03797j
Return to citation in text: [1] -
Bolm, C.; Hernández, J. G. Angew. Chem., Int. Ed. 2019, 58, 3285–3299. doi:10.1002/anie.201810902
Return to citation in text: [1] -
Friščić, T.; Mottillo, C.; Titi, H. M. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. doi:10.1002/anie.201906755
Return to citation in text: [1] -
Egorov, I. N.; Santra, S.; Kopchuk, D. S.; Kovalev, I. S.; Zyryanov, G. V.; Majee, A.; Ranu, B. C.; Rusinov, V. L.; Chupakhin, O. N. Green Chem. 2020, 22, 302–315. doi:10.1039/c9gc03414e
Return to citation in text: [1] -
Kubota, K.; Ito, H. Trends Chem. 2020, 2, 1066–1081. doi:10.1016/j.trechm.2020.09.006
Return to citation in text: [1] -
Pickhardt, W.; Grätz, S.; Borchardt, L. Chem. – Eur. J. 2020, 26, 12903–12911. doi:10.1002/chem.202001177
Return to citation in text: [1] -
Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. ChemSusChem 2022, 15, e202200362. doi:10.1002/cssc.202200362
Return to citation in text: [1] -
Colacino, E.; García, F. Mechanochemistry and Emerging Technologies for Sustainable Chemical Manufacturing; CRC Press: Boca Raton, FL, USA, 2023. doi:10.1201/9781003178187
Return to citation in text: [1] -
Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478
Return to citation in text: [1] -
Fantozzi, N.; Volle, J.-N.; Porcheddu, A.; Virieux, D.; García, F.; Colacino, E. Chem. Soc. Rev. 2023, 52, 6680–6714. doi:10.1039/d2cs00997h
Return to citation in text: [1] -
Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887
Return to citation in text: [1] -
Hutchings, B. P.; Crawford, D. E.; Gao, L.; Hu, P.; James, S. L. Angew. Chem., Int. Ed. 2017, 56, 15252–15256. doi:10.1002/anie.201706723
Return to citation in text: [1] -
Pladevall, B. S.; de Aguirre, A.; Maseras, F. ChemSusChem 2021, 14, 2763–2768. doi:10.1002/cssc.202100497
Return to citation in text: [1] -
Ardila-Fierro, K. J.; Hernández, J. G. Angew. Chem., Int. Ed. 2024, 63, e202317638. doi:10.1002/anie.202317638
Return to citation in text: [1] -
Zholdassov, Y. S.; Kwok, R. W.; Shlain, M. A.; Patel, M.; Marianski, M.; Braunschweig, A. B. RSC Mechanochem. 2024, 1, 11–32. doi:10.1039/d3mr00018d
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788
Return to citation in text: [1] -
Britton, R.; Gouverneur, V.; Lin, J.-H.; Meanwell, M.; Ni, C.; Pupo, G.; Xiao, J.-C.; Hu, J. Nat. Rev. Methods Primers 2021, 1, 47. doi:10.1038/s43586-021-00042-1
Return to citation in text: [1] -
Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639
Return to citation in text: [1] -
Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2021, 32, 3342–3354. doi:10.1016/j.cclet.2021.05.042
Return to citation in text: [1] -
Sap, J. B. I.; Meyer, C. F.; Straathof, N. J. W.; Iwumene, N.; am Ende, C. W.; Trabanco, A. A.; Gouverneur, V. Chem. Soc. Rev. 2021, 50, 8214–8247. doi:10.1039/d1cs00360g
Return to citation in text: [1] -
Fuchibe, K.; Ichikawa, J. Chem. Commun. 2023, 59, 2532–2540. doi:10.1039/d2cc03950h
Return to citation in text: [1] [2] -
Xie, Q.; Hu, J. Acc. Chem. Res. 2024, 57, 693–713. doi:10.1021/acs.accounts.3c00719
Return to citation in text: [1] -
Li, L.; Ni, C.; Xie, Q.; Hu, M.; Wang, F.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 9971–9975. doi:10.1002/anie.201705734
Return to citation in text: [1] [2] -
Olsen, D. A.; Osteraas, A. J. J. Appl. Polym. Sci. 1969, 13, 1523–1535. doi:10.1002/app.1969.070130715
Return to citation in text: [1] [2] -
Kirmse, W. Carbene Chemistry; Academic Press: New York, NY, USA, 1971; Vol. 1, pp 9–84. doi:10.1016/b978-0-12-409956-2.50006-6
Return to citation in text: [1] [2] -
van Bonn, P.; Ke, J.; Weike, C.; Ward, J. S.; Rissanen, K.; Bolm, C. CCS Chem. 2023, 5, 1737–1744. doi:10.31635/ccschem.023.202302783
Return to citation in text: [1] -
Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. J. Med. Chem. 2017, 60, 797–804. doi:10.1021/acs.jmedchem.6b01691
Return to citation in text: [1] -
Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604
Return to citation in text: [1] -
Lin, J.-H.; Xiao, J.-C. Extension to the Construction of ORf Motifs (OCF2H, OCFH2, OCH2CF3, OCFHCH3). In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D.; Ma, J.-A., Eds.; Wiley-VCH: Weinheim, Germany, 2020; Vol. 2, pp 267–288.
Return to citation in text: [1] -
Loison, A.; Toulgoat, F.; Billard, T.; Hanquet, G.; Panossian, A.; Leroux, F. R. Tetrahedron 2021, 99, 132458. doi:10.1016/j.tet.2021.132458
Return to citation in text: [1] -
Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 3206–3210. doi:10.1002/anie.201611823
Return to citation in text: [1] [2] -
Ford, J.; Hopkin, B.; Sap, J. B. I.; Gouverneur, V. Isr. J. Chem. 2023, 63, e202300109. doi:10.1002/ijch.202300109
Return to citation in text: [1] -
Liu, G.; Wang, X.; Xu, X.-H.; Lu, X.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Org. Lett. 2013, 15, 1044–1047. doi:10.1021/ol4000313
Return to citation in text: [1] -
Lin, X.; Weng, Z. Org. Biomol. Chem. 2015, 13, 3432–3437. doi:10.1039/c5ob00020c
Return to citation in text: [1] -
Liu, C.; Deng, X.-Y.; Zeng, X.-L.; Zhao, G.; Lin, J.-H.; Wang, H.; Xiao, J.-C. J. Fluorine Chem. 2016, 192, 27–30. doi:10.1016/j.jfluchem.2016.10.011
Return to citation in text: [1] -
Yue, C.-B.; Lin, J.-H.; Cai, J.; Zhang, C.-P.; Zhao, G.; Xiao, J.-C.; Li, H. RSC Adv. 2016, 6, 35705–35708. doi:10.1039/c6ra06338a
Return to citation in text: [1] -
Liu, G.-K.; Li, X.; Qin, W.-B.; Lin, W.-F.; Lin, L.-T.; Chen, J.-Y.; Liu, J.-J. Chin. Chem. Lett. 2019, 30, 1515–1518. doi:10.1016/j.cclet.2019.03.036
Return to citation in text: [1] -
Cai, X.; Zhai, Y.; Ghiviriga, I.; Abboud, K. A.; Dolbier, W. R. J. Org. Chem. 2004, 69, 4210–4215. doi:10.1021/jo049570y
Return to citation in text: [1] -
Cai, X.; Wu, K.; Dolbier, W. R., Jr. J. Fluorine Chem. 2005, 126, 477–480. doi:10.1016/j.jfluchem.2004.11.006
Return to citation in text: [1] -
Fuchibe, K.; Koseki, Y.; Sasagawa, H.; Ichikawa, J. Chem. Lett. 2011, 40, 1189–1191. doi:10.1246/cl.2011.1189
Return to citation in text: [1] -
Fuchibe, K.; Koseki, Y.; Aono, T.; Sasagawa, H.; Ichikawa, J. J. Fluorine Chem. 2012, 133, 52–60. doi:10.1016/j.jfluchem.2011.09.012
Return to citation in text: [1] -
Jia, Y.; Yuan, Y.; Huang, J.; Jiang, Z.-X.; Yang, Z. Org. Lett. 2021, 23, 2670–2675. doi:10.1021/acs.orglett.1c00577
Return to citation in text: [1]
| 39. | Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 3206–3210. doi:10.1002/anie.201611823 |
| 50. | Jia, Y.; Yuan, Y.; Huang, J.; Jiang, Z.-X.; Yang, Z. Org. Lett. 2021, 23, 2670–2675. doi:10.1021/acs.orglett.1c00577 |
| 1. | Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c |
| 2. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a |
| 3. | Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a |
| 4. | Andersen, J.; Mack, J. Green Chem. 2018, 20, 1435–1443. doi:10.1039/c7gc03797j |
| 5. | Bolm, C.; Hernández, J. G. Angew. Chem., Int. Ed. 2019, 58, 3285–3299. doi:10.1002/anie.201810902 |
| 6. | Friščić, T.; Mottillo, C.; Titi, H. M. Angew. Chem., Int. Ed. 2020, 59, 1018–1029. doi:10.1002/anie.201906755 |
| 7. | Egorov, I. N.; Santra, S.; Kopchuk, D. S.; Kovalev, I. S.; Zyryanov, G. V.; Majee, A.; Ranu, B. C.; Rusinov, V. L.; Chupakhin, O. N. Green Chem. 2020, 22, 302–315. doi:10.1039/c9gc03414e |
| 8. | Kubota, K.; Ito, H. Trends Chem. 2020, 2, 1066–1081. doi:10.1016/j.trechm.2020.09.006 |
| 9. | Pickhardt, W.; Grätz, S.; Borchardt, L. Chem. – Eur. J. 2020, 26, 12903–12911. doi:10.1002/chem.202001177 |
| 10. | Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. ChemSusChem 2022, 15, e202200362. doi:10.1002/cssc.202200362 |
| 11. | Colacino, E.; García, F. Mechanochemistry and Emerging Technologies for Sustainable Chemical Manufacturing; CRC Press: Boca Raton, FL, USA, 2023. doi:10.1201/9781003178187 |
| 19. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 20. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 21. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 22. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 23. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 24. | Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788 |
| 25. | Britton, R.; Gouverneur, V.; Lin, J.-H.; Meanwell, M.; Ni, C.; Pupo, G.; Xiao, J.-C.; Hu, J. Nat. Rev. Methods Primers 2021, 1, 47. doi:10.1038/s43586-021-00042-1 |
| 26. | Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639 |
| 27. | Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2021, 32, 3342–3354. doi:10.1016/j.cclet.2021.05.042 |
| 29. | Fuchibe, K.; Ichikawa, J. Chem. Commun. 2023, 59, 2532–2540. doi:10.1039/d2cc03950h |
| 48. | Fuchibe, K.; Koseki, Y.; Sasagawa, H.; Ichikawa, J. Chem. Lett. 2011, 40, 1189–1191. doi:10.1246/cl.2011.1189 |
| 49. | Fuchibe, K.; Koseki, Y.; Aono, T.; Sasagawa, H.; Ichikawa, J. J. Fluorine Chem. 2012, 133, 52–60. doi:10.1016/j.jfluchem.2011.09.012 |
| 15. | Hutchings, B. P.; Crawford, D. E.; Gao, L.; Hu, P.; James, S. L. Angew. Chem., Int. Ed. 2017, 56, 15252–15256. doi:10.1002/anie.201706723 |
| 16. | Pladevall, B. S.; de Aguirre, A.; Maseras, F. ChemSusChem 2021, 14, 2763–2768. doi:10.1002/cssc.202100497 |
| 17. | Ardila-Fierro, K. J.; Hernández, J. G. Angew. Chem., Int. Ed. 2024, 63, e202317638. doi:10.1002/anie.202317638 |
| 18. | Zholdassov, Y. S.; Kwok, R. W.; Shlain, M. A.; Patel, M.; Marianski, M.; Braunschweig, A. B. RSC Mechanochem. 2024, 1, 11–32. doi:10.1039/d3mr00018d |
| 31. | Li, L.; Ni, C.; Xie, Q.; Hu, M.; Wang, F.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 9971–9975. doi:10.1002/anie.201705734 |
| 32. | Olsen, D. A.; Osteraas, A. J. J. Appl. Polym. Sci. 1969, 13, 1523–1535. doi:10.1002/app.1969.070130715 |
| 33. | Kirmse, W. Carbene Chemistry; Academic Press: New York, NY, USA, 1971; Vol. 1, pp 9–84. doi:10.1016/b978-0-12-409956-2.50006-6 |
| 3. | Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a |
| 14. | Hernández, J. G.; Bolm, C. J. Org. Chem. 2017, 82, 4007–4019. doi:10.1021/acs.joc.6b02887 |
| 41. | Liu, G.; Wang, X.; Xu, X.-H.; Lu, X.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Org. Lett. 2013, 15, 1044–1047. doi:10.1021/ol4000313 |
| 42. | Lin, X.; Weng, Z. Org. Biomol. Chem. 2015, 13, 3432–3437. doi:10.1039/c5ob00020c |
| 43. | Liu, C.; Deng, X.-Y.; Zeng, X.-L.; Zhao, G.; Lin, J.-H.; Wang, H.; Xiao, J.-C. J. Fluorine Chem. 2016, 192, 27–30. doi:10.1016/j.jfluchem.2016.10.011 |
| 44. | Yue, C.-B.; Lin, J.-H.; Cai, J.; Zhang, C.-P.; Zhao, G.; Xiao, J.-C.; Li, H. RSC Adv. 2016, 6, 35705–35708. doi:10.1039/c6ra06338a |
| 45. | Liu, G.-K.; Li, X.; Qin, W.-B.; Lin, W.-F.; Lin, L.-T.; Chen, J.-Y.; Liu, J.-J. Chin. Chem. Lett. 2019, 30, 1515–1518. doi:10.1016/j.cclet.2019.03.036 |
| 12. | Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478 |
| 13. | Fantozzi, N.; Volle, J.-N.; Porcheddu, A.; Virieux, D.; García, F.; Colacino, E. Chem. Soc. Rev. 2023, 52, 6680–6714. doi:10.1039/d2cs00997h |
| 46. | Cai, X.; Zhai, Y.; Ghiviriga, I.; Abboud, K. A.; Dolbier, W. R. J. Org. Chem. 2004, 69, 4210–4215. doi:10.1021/jo049570y |
| 47. | Cai, X.; Wu, K.; Dolbier, W. R., Jr. J. Fluorine Chem. 2005, 126, 477–480. doi:10.1016/j.jfluchem.2004.11.006 |
| 34. | van Bonn, P.; Ke, J.; Weike, C.; Ward, J. S.; Rissanen, K.; Bolm, C. CCS Chem. 2023, 5, 1737–1744. doi:10.31635/ccschem.023.202302783 |
| 39. | Xie, Q.; Ni, C.; Zhang, R.; Li, L.; Rong, J.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 3206–3210. doi:10.1002/anie.201611823 |
| 32. | Olsen, D. A.; Osteraas, A. J. J. Appl. Polym. Sci. 1969, 13, 1523–1535. doi:10.1002/app.1969.070130715 |
| 33. | Kirmse, W. Carbene Chemistry; Academic Press: New York, NY, USA, 1971; Vol. 1, pp 9–84. doi:10.1016/b978-0-12-409956-2.50006-6 |
| 40. | Ford, J.; Hopkin, B.; Sap, J. B. I.; Gouverneur, V. Isr. J. Chem. 2023, 63, e202300109. doi:10.1002/ijch.202300109 |
| 31. | Li, L.; Ni, C.; Xie, Q.; Hu, M.; Wang, F.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 9971–9975. doi:10.1002/anie.201705734 |
| 28. | Sap, J. B. I.; Meyer, C. F.; Straathof, N. J. W.; Iwumene, N.; am Ende, C. W.; Trabanco, A. A.; Gouverneur, V. Chem. Soc. Rev. 2021, 50, 8214–8247. doi:10.1039/d1cs00360g |
| 29. | Fuchibe, K.; Ichikawa, J. Chem. Commun. 2023, 59, 2532–2540. doi:10.1039/d2cc03950h |
| 30. | Xie, Q.; Hu, J. Acc. Chem. Res. 2024, 57, 693–713. doi:10.1021/acs.accounts.3c00719 |
| 35. | Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. J. Med. Chem. 2017, 60, 797–804. doi:10.1021/acs.jmedchem.6b01691 |
| 36. | Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604 |
| 37. | Lin, J.-H.; Xiao, J.-C. Extension to the Construction of ORf Motifs (OCF2H, OCFH2, OCH2CF3, OCFHCH3). In Emerging Fluorinated Motifs: Synthesis, Properties, and Applications; Cahard, D.; Ma, J.-A., Eds.; Wiley-VCH: Weinheim, Germany, 2020; Vol. 2, pp 267–288. |
| 38. | Loison, A.; Toulgoat, F.; Billard, T.; Hanquet, G.; Panossian, A.; Leroux, F. R. Tetrahedron 2021, 99, 132458. doi:10.1016/j.tet.2021.132458 |
© 2024 Ke et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.